This guide to the modules on the Region page is illustrated in the Common Metabolic Diseases Knowledge Portal (CMDKP) but applies to all Knowledge Portals that display genetic and genomic results.
Click on a link to see documentation for that section of the Region page:
Most significant variant associations in the region
You can navigate to the Region page by entering a gene name or the coordinates of a genomic region into the search box on the portal home page and clicking Search region. If you enter a gene name, the region shown is defined as the coding sequence of the gene plus 50kb of upstream and downstream flanking sequences.
Set page level parameters
The gray box at the top of the Region page allows you to navigate to a different Portal page or to select an ancestry for the associations viewed on the page.
- The Begin new search box allows you to navigate to a different Gene, Variant, Region, Phenotype, or Tissue page. Begin typing in the box and select an option from the list.
- The Set Ancestry menu allows you to select an ancestry and change the Region page to display only associations from that ancestry. Note that associations are not available for every ancestry in every region.
Region coordinates
This section displays the coordinates of the region that is currently displayed:
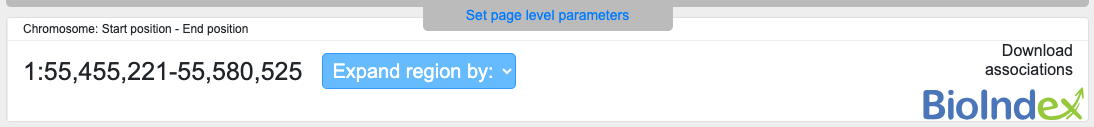
All coordinates are in the hg19 (GRCh37) genome build.
Click the Expand region menu to expand the region by 50, 100, or 150kb both up- and downstream:
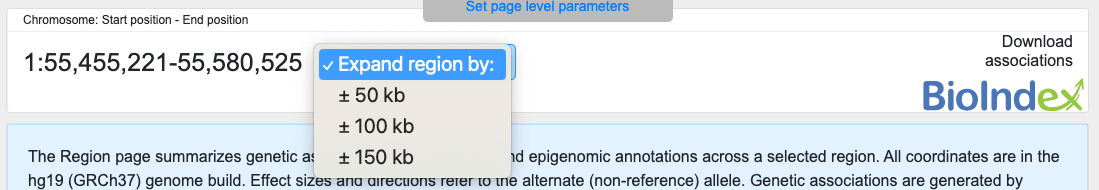
On the right is a link to the HuGeAMP BioIndex, which offers programmatic access to all the data in the Knowledge Portals.
Page description
The page description section summarizes the content of the page, links to workflows and documentation, and states three important parameters for the results displayed on the page (and throughout the portal):
- Coordinates are in the hg19 (GRCh37) genome build
- Effect sizes and directions refer to the alternate (non-reference) allele
- Genetic associations are generated by bottom-line integrative analysis

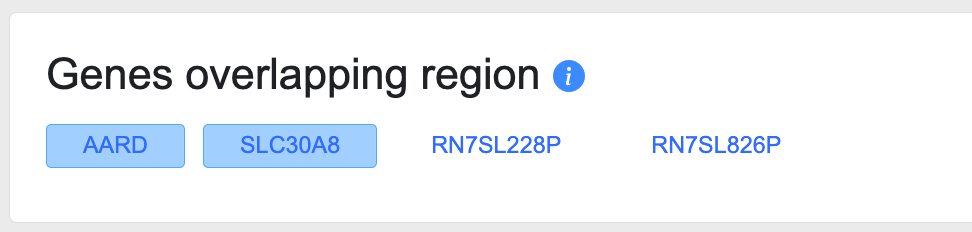
Genes overlapping region
This section shows the genes whose coding sequences are located (completely or partially) within the region spanned by the page. Clicking on a gene name navigates you to its Gene page.

In this section, protein-coding genes are shown in solid blue boxes, while non-coding RNA genes are not displayed in boxes:
Most significant variant associations in the region
The top of this section displays a Phenome-wide Association (PheWAS) plot showing the most significant associations in the region for the phenotypes available in the portal you are viewing. See this page for an overview of the phenotypes available in each portal. Try the new Association to Function Knowledge Portal to view associations for all phenotypes or set your own custom phenotype view.
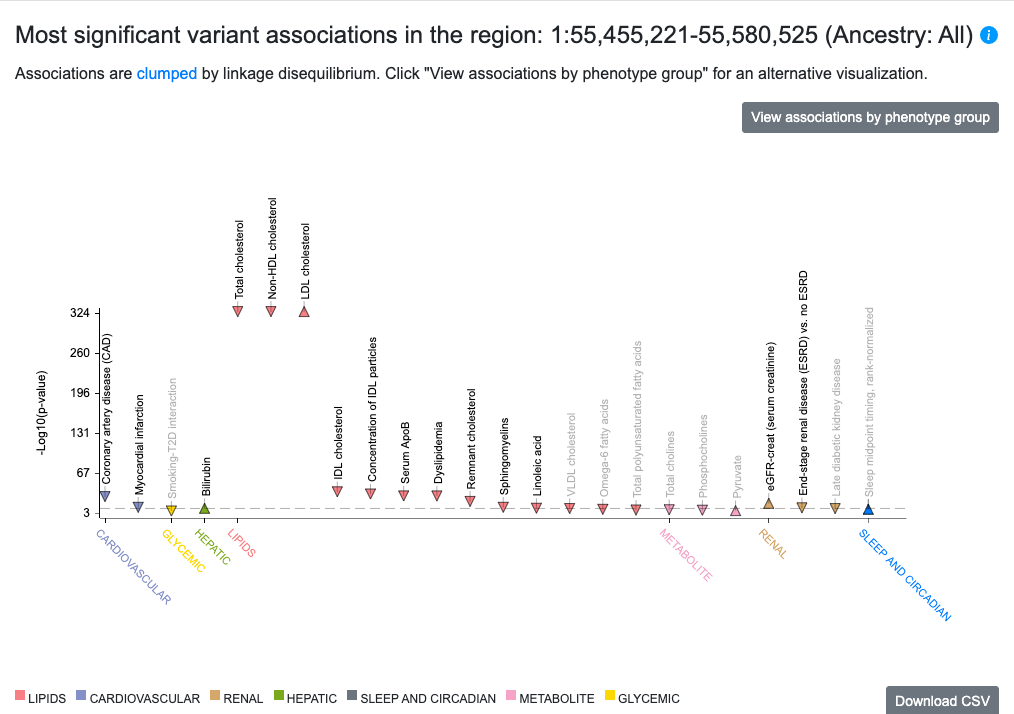
The points on the plot are colored by phenotype group. Upward-facing or downward-facing triangles indicate positive or negative direction of effect, respectively. Mouse over a point to see a tooltip with p-value and beta for the association. If you click the tooltip, you have the options to 1) add the phenotype to the page, which adds a LocusZoom plot for that phenotype and adds associations for that phenotype to the Variants in region table below; or 2) navigate to the Phenotype page for that phenotype.
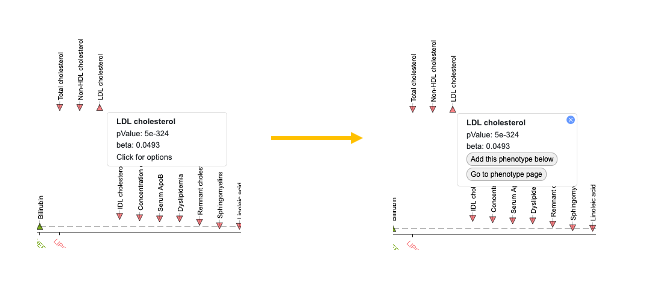
At the upper right of the PheWAS plot, the View associations by phenotype group button leads to an alternative view of associations across the region:
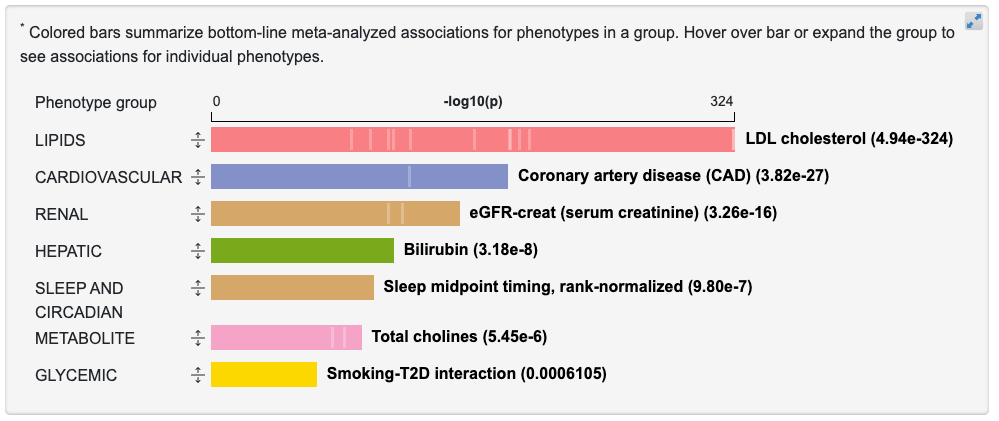
Each bar represents the associations for phenotypes in a broad category. The length of the bar is proportional to the negative log10 of the most significant p-value for a phenotype in that category. If you mouse over the vertical lines within each bar, a tooltip shows associations for individual phenotypes within that group:
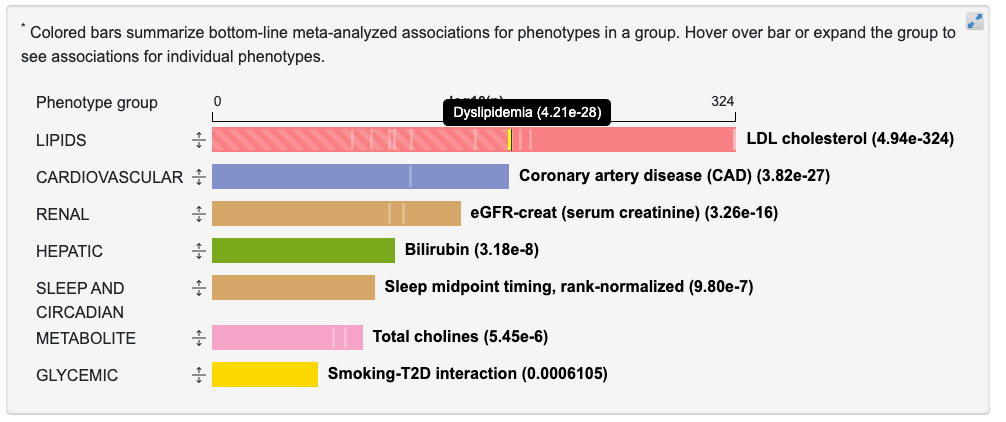
Clicking the arrow icon to the left of a bar expands it to show associations for the individual phenotypes within that group:
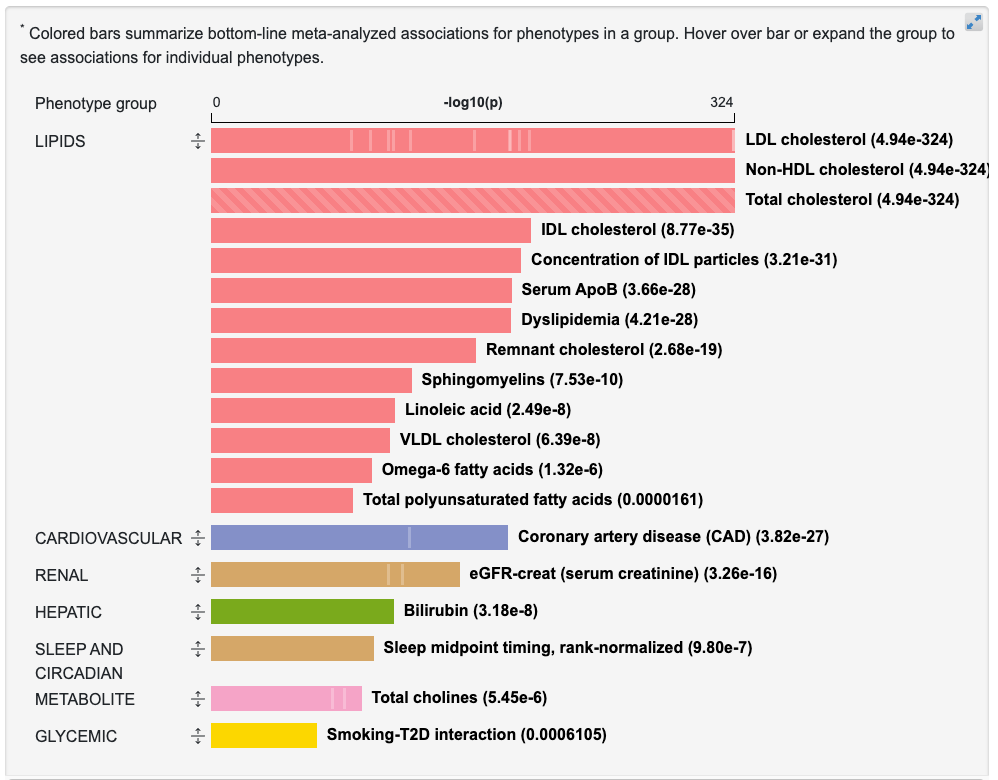
You can return to the PheWAS plot by clicking View associations by individual phenotype on the right above the graphic.
The table below the graphics shows LD-clumped associations within this region that either have genome-wide significance or are in the top 99th percentile of all associations for that trait across the genome. The lead variant in each clump is shown by default:
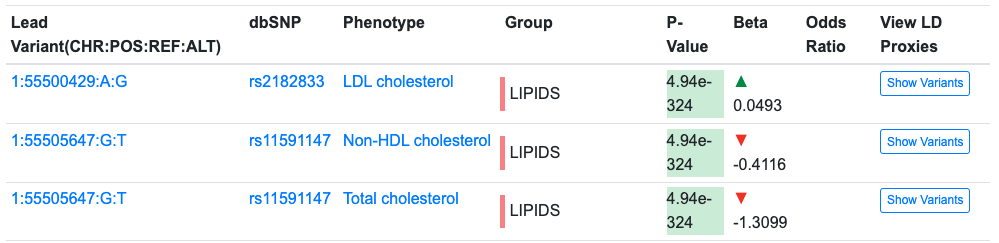
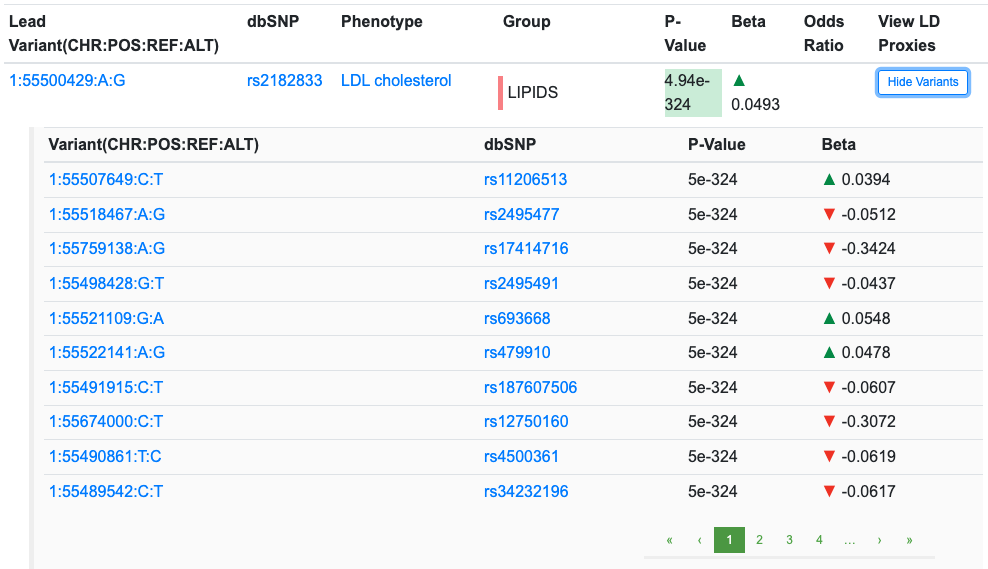
The Download CSV button at the upper right of the table download a file of the lead variants and their associations. Clicking the Show Variants button displays a list of the individual variants in a clump:
Genomic Region Miner (GEM)
The GEM module allows you to visualize genetic associations for multiple phenotypes across a region. By default, GEM shows a LocusZoom plot of the bottom-line genetic associations for the most significantly associated phenotype in the region. Click Save PNG to download a publication-quality image of the plot.
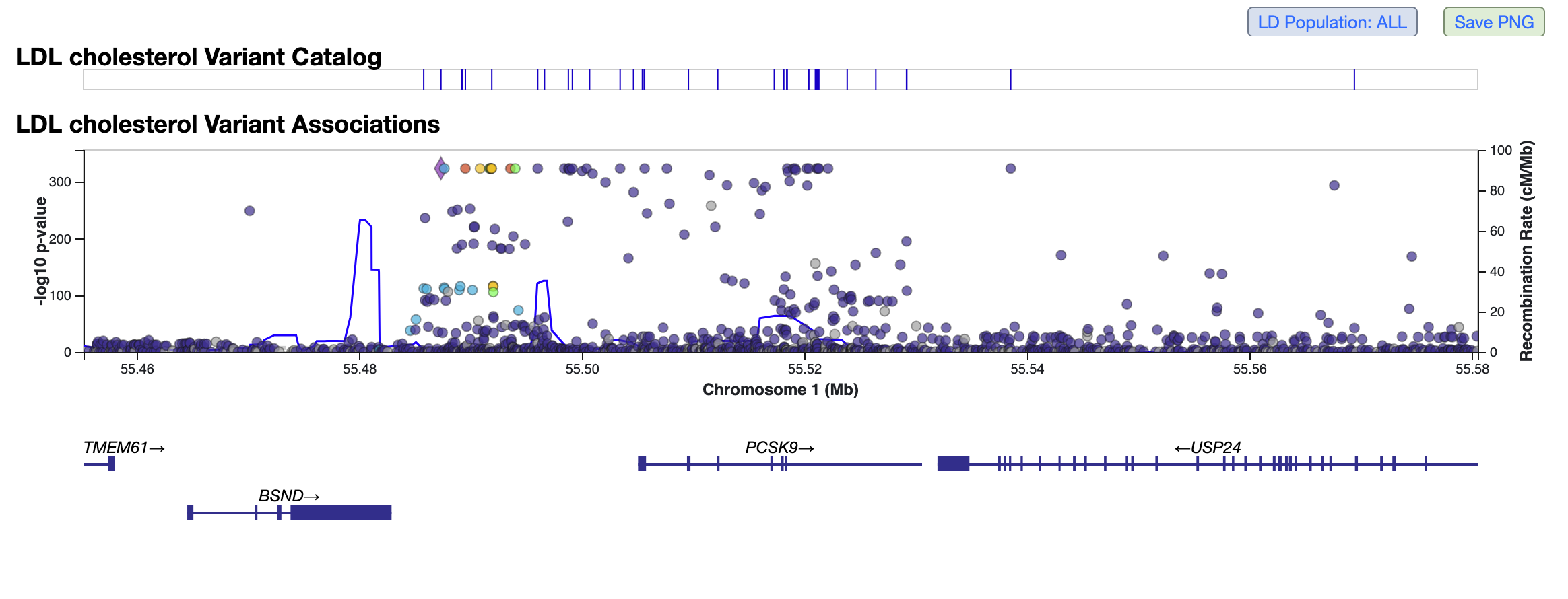
To zoom in or out on most operating systems, hold the Shift key while scrolling; on a MacBook running Linux, hold the Option key while scrolling. Pan left or right by clicking and dragging. Mouse over a point to see variant information and p-values. Points are colored by LD; click Show legend to see the values corresponding to different colors. Click the LD Population button to set the LD population to a specific ancestry. You can set a different LD reference variant in the plot by mousing over a point and clicking Make LD Reference in the tooltip.
Above the plot, a tick mark on the the GWAS Catalog track indicates whether a variant at that genomic position has an association of genome-wide significance or greater (p < 5 x 10e-8), for any phenotype, that is noted in the GWAS Catalog. Note that the GWAS catalog specifies the risk allele, but not the non-risk allele, so it is important to consult the publication for a variant to determine which alleles were used when testing for association.
Below the plot, click on a gene to for links to its records in the Ensembl and gnomAD databases.
The associations shown in the LocusZoom plot are listed in the Variants in Region table below:
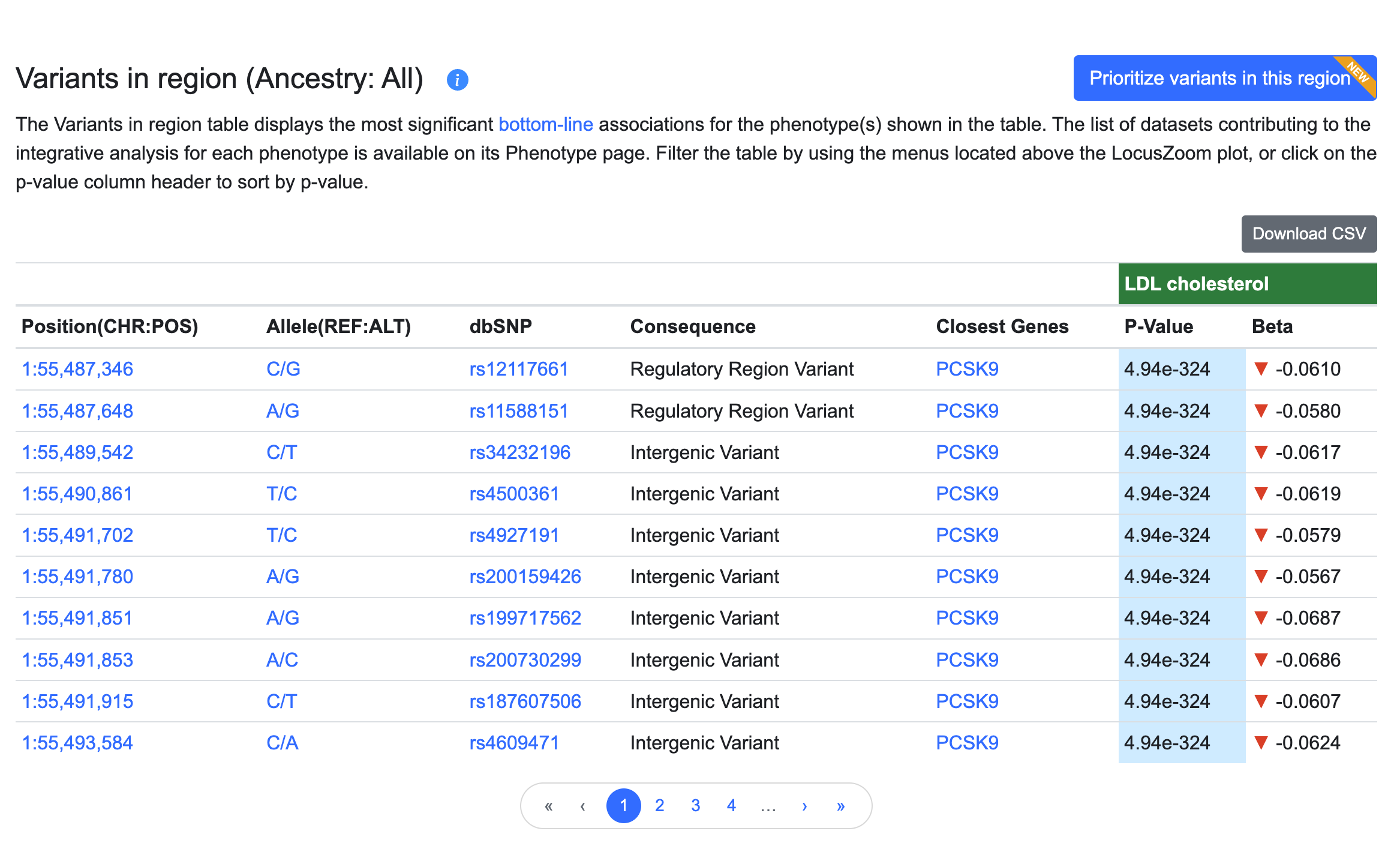
In the table:
- the Position column indicates the chromosome and coordinate of the variant (hg19 genome build), and is linked to a Region page centered on the variant.
- Allele shows reference allele/alternate allele and is linked to the Variant page.
- the dbSNP column shows the reference SNP number (rsID) assigned to this variant by dbSNP and is linked to the Variant page.
- the Consequence column shows the impact of sequence variation at this position for overlapping genes or transcripts, as predicted by the Ensembl Variant Effect Predictor (VEP) and expressed using Sequence Ontology terms.
- the Closest genes column indicates the one or more genes nearest to the variant, as predicted by VEP. Gene names are linked to their Gene pages.
- the p-value column represents the probability that the observed frequency difference would occur by chance. p-values in this table are bottom-line p-values derived from integrative analysis.
- Odds ratios (OR) are reported for dichotomous (binary) traits. OR > 1 indicates a positive correlation between the effect allele and a phenotype, while an OR < 1 indicates a negative correlation. OR refers to the alternate (non-reference) allele.
- Beta coefficients are reported for reported for continuous (quantitative) traits. Beta is the estimated difference in phenotype between a heterozygous carrier of an effect allele and a homozygous reference allele carrier, shown in terms of the standard deviation of the phenotype (For a few traits that are not originally reported in terms of S.D. units, we approximate the units by a regression of the standard errors.) A beta coefficient > 0 indicates a positive correlation between the effect allele and a phenotype, while a beta < 0 indicates a negative correlation. Beta refers to the alternate (non-reference) allele.
You can add LocusZoom plots for more phenotypes using the Add phenotypes menu:
Begin typing a phenotype, then select one from the menu. When you add a plot, the associations for that phenotype are also listed in the Variants in region table.
You can filter the associations displayed in the LocusZoom track(s) and the table using the Filter plots and table menus above the plot:
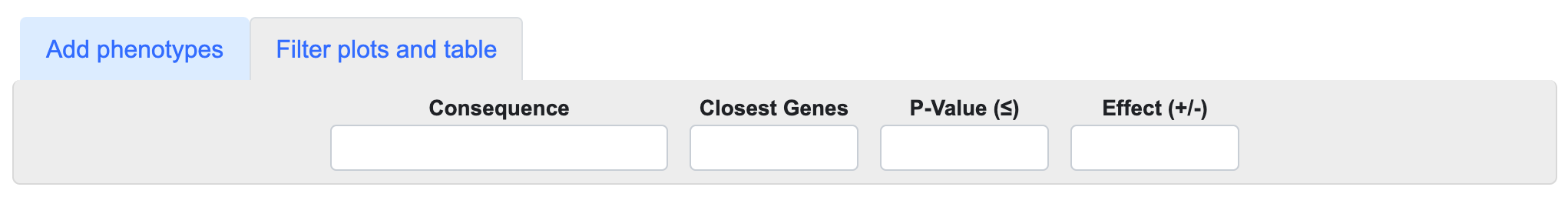
- Begin typing in the Consequence or Closest Genes boxes to select one or more variant consequences and/or closest genes as filtering criteria for the table and plot.
- Enter a p-value in the format "5e-8" to set an upper threshold for the p-values of the associations displayed.
- Enter "positive" or "negative" into the Effect box to filter by association direction of effect.