This guide to the modules on the Variant page is illustrated in the Common Metabolic Diseases Knowledge Portal (CMDKP) but applies to all Knowledge Portals that display genetic and genomic results.
The Variant Page summarizes impact predictions and genetic associations for a single variant. Click on a link to see documentation for that section of the Variant page:
Predicted variant consequences
Transcription factor binding motifs
You can navigate to a Variant page by entering a variant ID into the search box on the portal home page or by clicking on a variant ID anywhere it appears on the portal.
Set page level parameters
The gray box at the top of the Variant page allows you to navigate to a different Portal page or to select an ancestry for the associations viewed on the page.
- The Begin new search box allows you to navigate to a different Gene, Variant, Region, Phenotype, or Tissue page. Begin typing in the box and select an option from the list.
- Ancestry: Click the menu to select an ancestry and change the Variant page to display only associations from that ancestry. Note that associations are not available for every ancestry in every region.
Variant IDs
This section displays the rsID of the variant as well as a variant ID in the format Chromosome:Position (hg19 (GRCh37) genome build):Reference allele:Alternate allele.
Click the Explore region button to go to a Region page spanning 100 kb and centered on the variant.
Page description
The page description section summarizes the content of the page and links to workflows and documentation.

Closest genes
This section shows the protein-coding gene that is nearest to the variant, as predicted by the Ensembl Variant Effect Predictor (VEP). Clicking on a gene name navigates you to its Gene page.

Predicted variant consequences
This section shows the predicted impact of the variant on transcription, splicing, and protein coding, as determined by VEP:
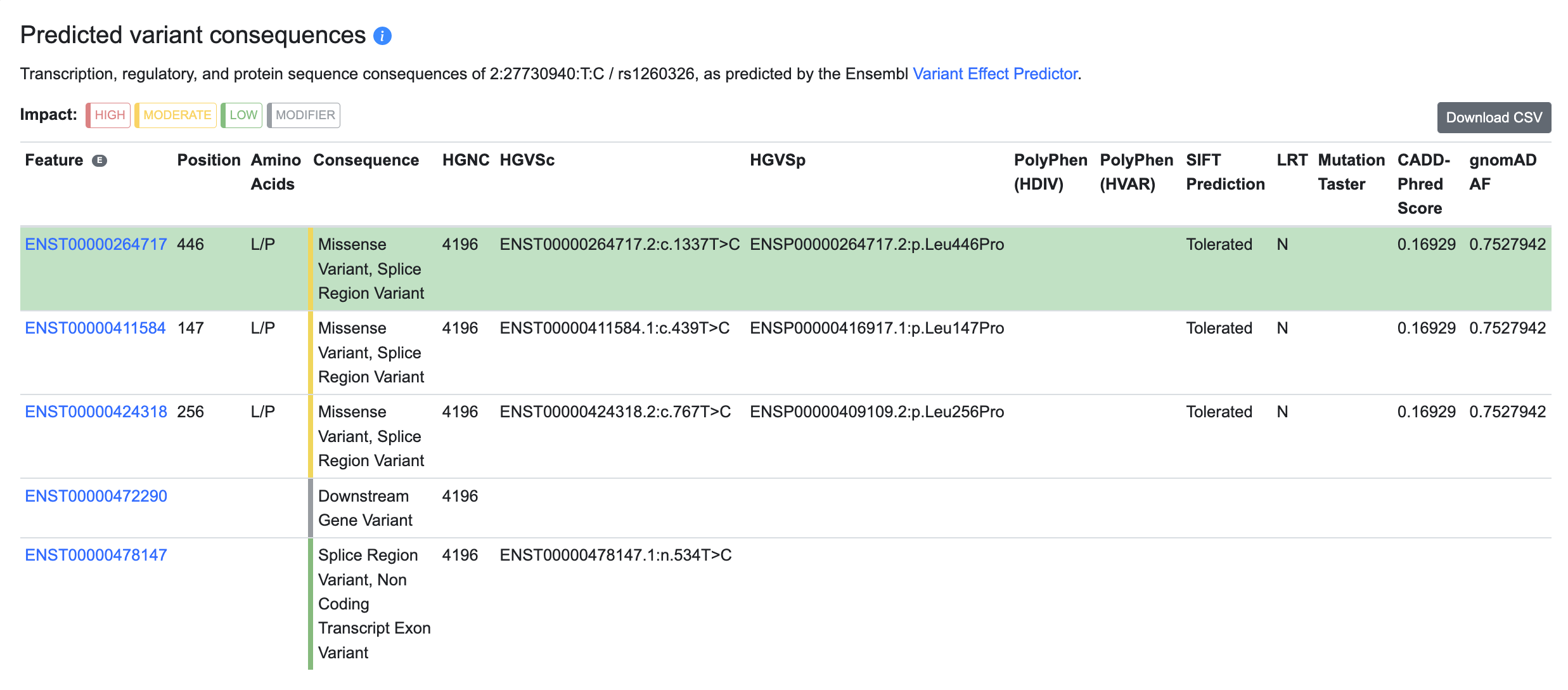
The row highlighted in green shows the most severe predicted consequence of the variant.
The columns of the table display:
- Feature: the Ensembl ID of the genomic feature in which the variant is located, linked to its Ensembl record
- Position: the amino acid position within the encoded protein, for variants that affect protein-coding sequences
- Amino acids: the amino acids encoded by the reference sequence and the variant sequence, for variants that affect protein-coding sequences
- Consequence: the consequence type of the variant, as expressed using Sequence Ontology terms
- HGNC: the HUGO Gene Nomenclature Committee (HGNC) identifier of the gene in which the variant is located or the nearest gene to the variant
- HGVSc: the identifier of the coding sequence of the gene in which the variant is located or the nearest gene to the variant, from the Human Genome Variation Society (HGVS)
- HGVSp: the identifier of the protein sequence encoded by the gene in which the variant is located or the nearest gene to the variant, from the Human Genome Variation Society (HGVS)
- PolyPhen (HDIV): prediction of deleteriousness of the variant by the PolyPhen HDIV method (Adzhubei et al., 2010)
- PolyPhen (HVAR): prediction of deleteriousness of the variant by the PolyPhen HVAR method (Adzhubei et al., 2010)
- SIFT prediction: prediction of deleteriousness of the variant by the Sorting Intolerant From Tolerant (SIFT) algorithm (Kumar et al, 2009)
- LRT: prediction of deleteriousness of the variant by the Likelihood Ratio Test (LRT; Chun and Fay, 2009)
- MutationTaster: prediction of deleteriousness of the variant by the MutationTaster method (Schwarz et al., 2010)
- CADD-Phred score: deleteriousness score of the variant as generated by the Combined Annotation Dependent Depletion tool CADD (Rentzsch et al., 2019)
- gnomAD AF: allele frequency of the variant, from the gnomAD database
The overall predicted impact of the variant is indicated by a colored bar between the Amino acids and Consequence columns, as shown below:
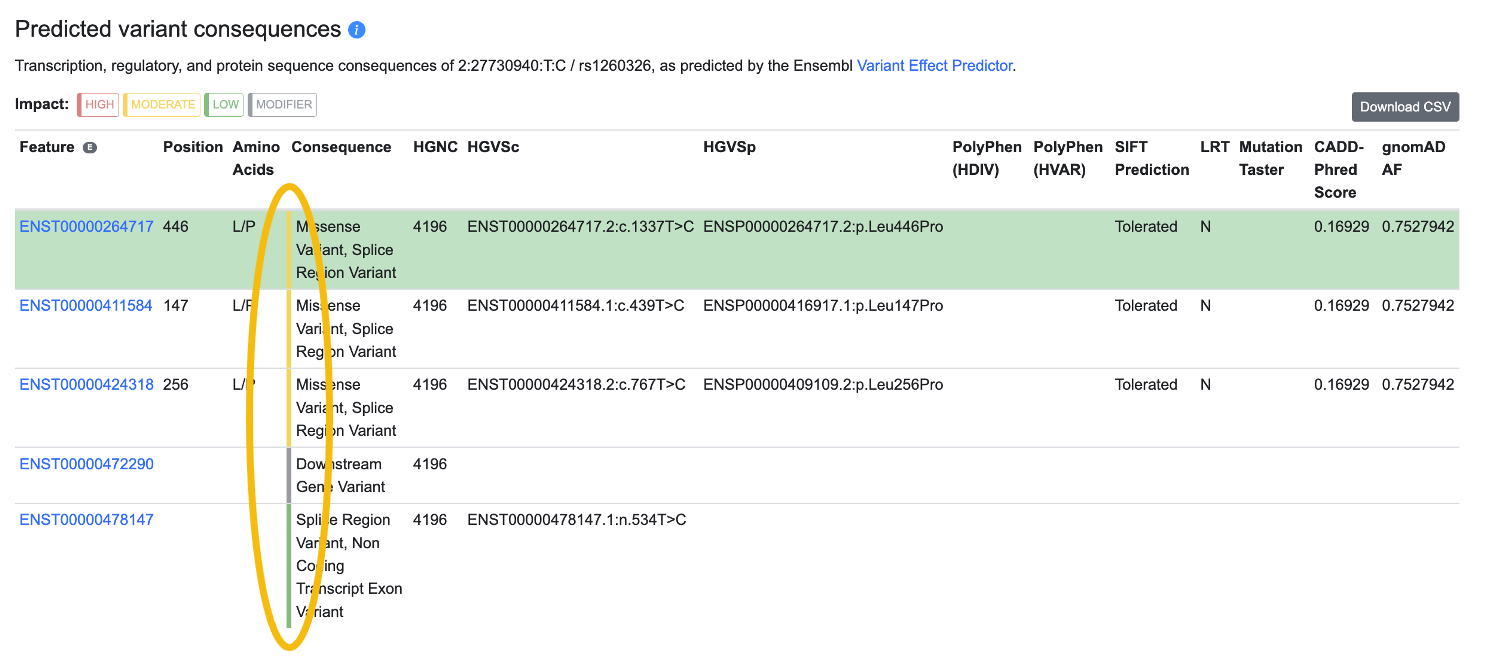
High-impact variants are indicated by a red bar; moderate-impact by yellow; low-impact by green; and modifier by grey. The Sequence Ontology annotations included in each category are listed on this page.
Variant PheWAS associations
This section displays a Phenome-wide Association (PheWAS) plot showing the most significant bottom-line associations (p ≤ 0.05) of the variant for the phenotypes available in the portal you are viewing. See this page for an overview of the phenotypes available in each portal. Try the new Association to Function Knowledge Portal to view associations for all phenotypes or set your own custom phenotype view.
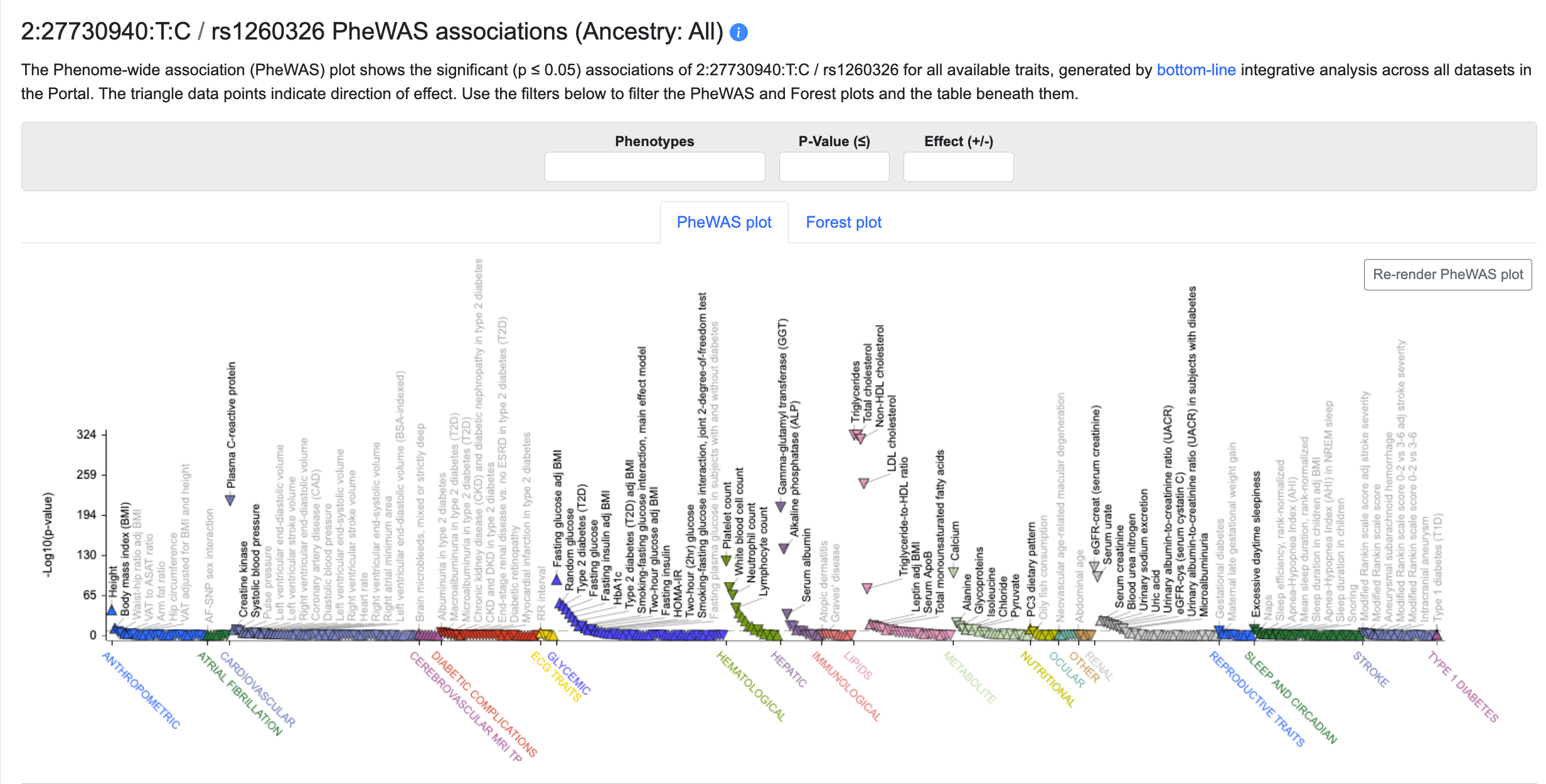
The points on the plot are colored by phenotype group. Upward-facing or downward-facing triangles indicate positive or negative direction of effect, respectively. Mouse over a point to see a tooltip with p-value and beta for the association.
The filters above the table allow you to customize the PheWAS display by 1) entering one or more phenotypes (one at a time) to view only the associations for those phenotypes; 2) set a threshold for the minimum p-value of the associations displayed; or 3) choose to view associations with only positive or negative direction of effect. Filtering the plot also filters the variants listed in the table below.
Click the Forest plot tab to view the same associations in a forest plot:
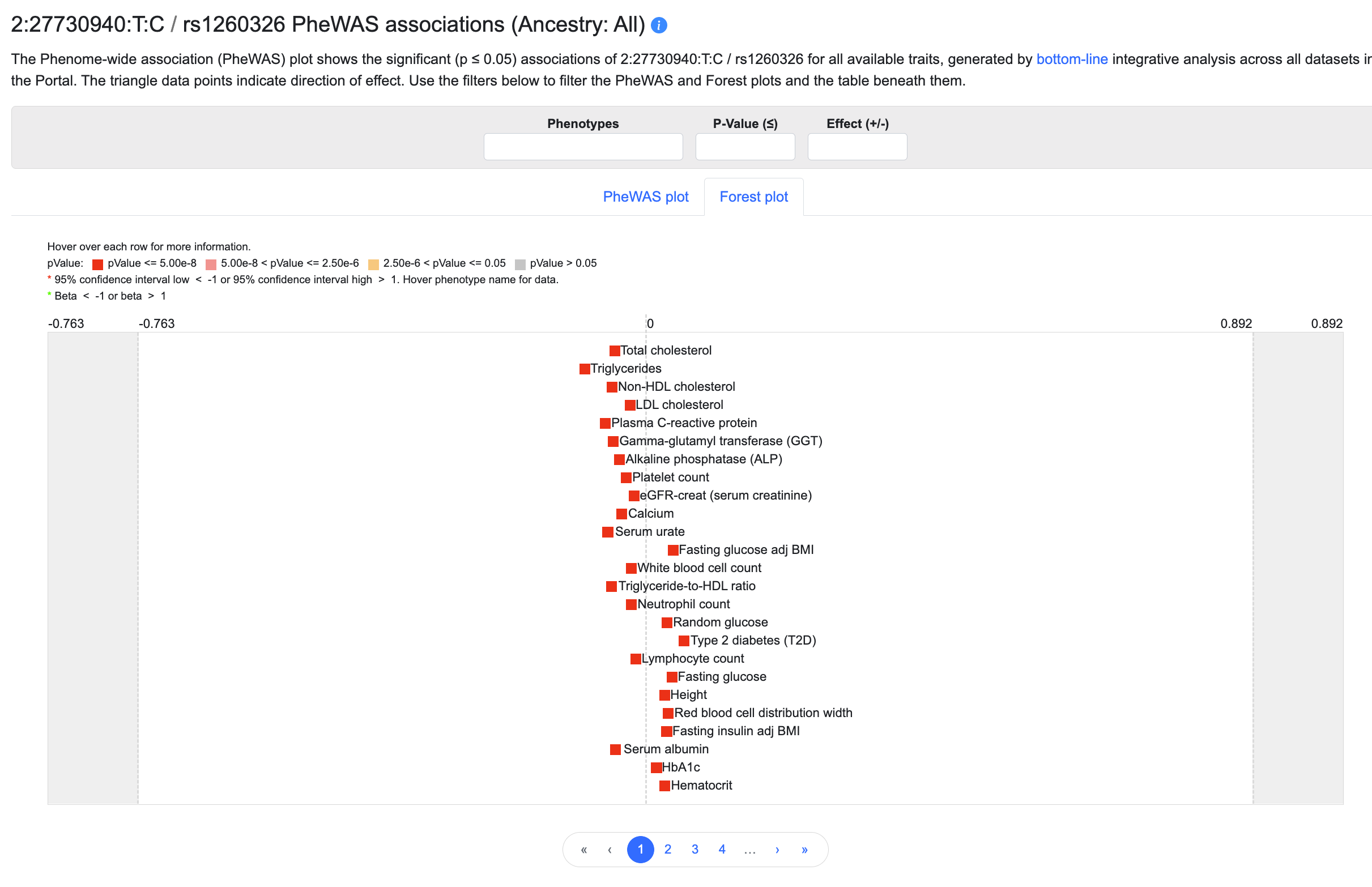
Mouse over the plot to see details about each association. The forest plot may be filtered in the same way as the PheWAS plot (see above).
In the forest plot, beta is shown in terms of the standard deviation of the phenotype. For traits that are not originally reported in terms of s.d. units, we approximate the units by a regression of the standard errors.
Below the plot, a downloadable table lists the variant associations displayed in the plot. In the table, effective sample size refers to the number of samples in a balanced case:control design that would yield equivalent power as the original study. It is mathematically defined as 4 / (1 / n.case + 1 / n.ctrl).
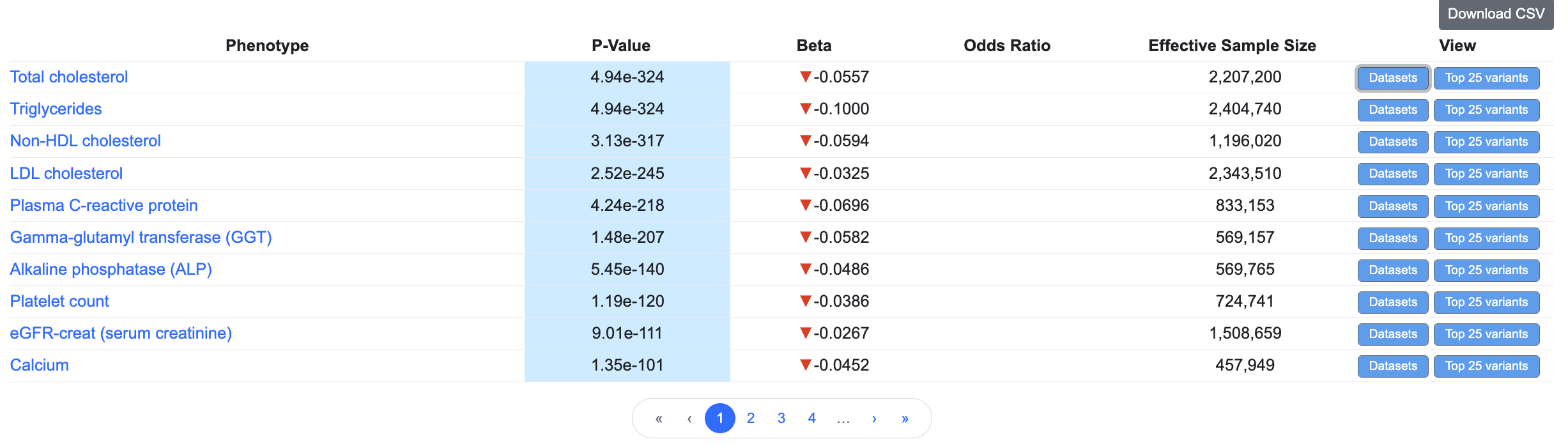
Click the Top 25 variants button to see the top 25 variants in the same LD-clump as the variant of the page:
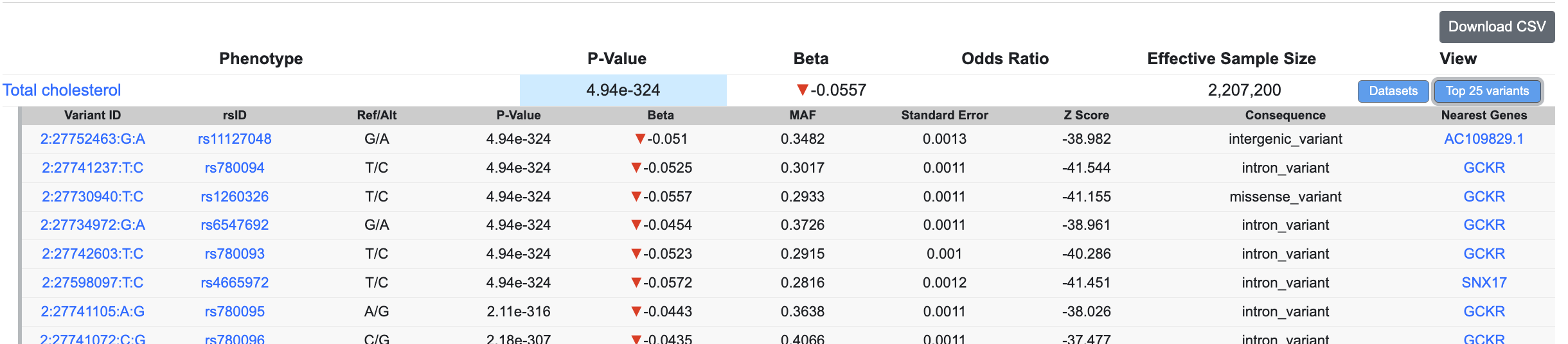
Click the View all variants in clump button (visible after clicking the Top 25 variants button) to see a plot and table illustrating the entire clump.
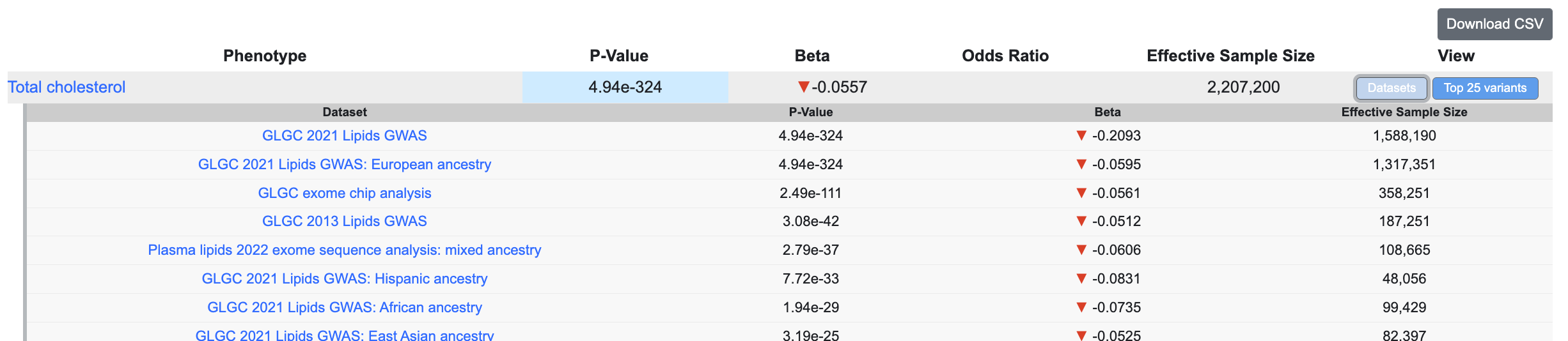
Click the Datasets button to see associations of the variant of the page in individual genetic association datasets:
Transcription factor binding motifs
This table displays predicted transcription factor (TF) binding motifs from Haploreg that overlap the position of the variant.
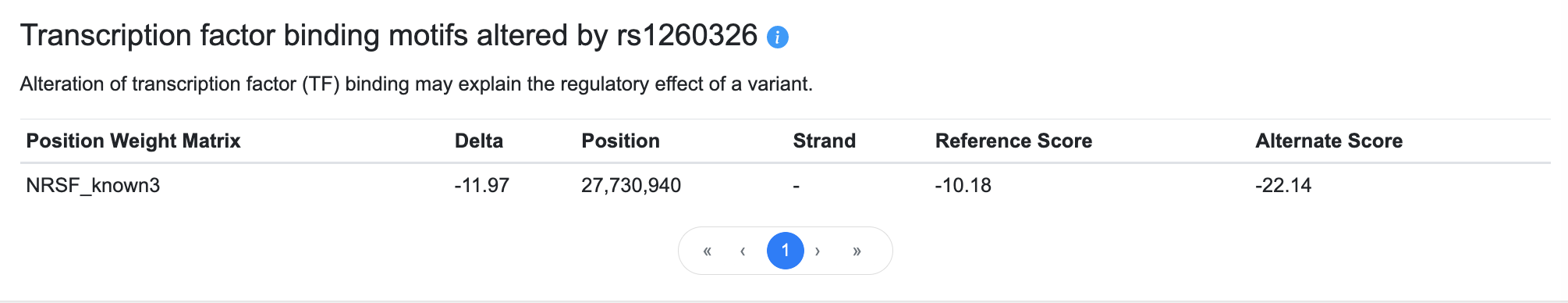
The columns of the table are:
- the Position weight matrix column indicates the identifier of the position weight matrix, or DNA sequence motif, that the variant is predicted to alter
- Delta indicates the difference between Reference and Alternate scores, representing the degree to which the TF binding motif is affected by the alternate allele
- Position shows the position of the variant within the TF binding motif
- Strand indicates the DNA strand (plus or minus) on which the TF binding motif is found
- Reference score represents the degree to which the reference allele affects the TF binding motif
- Alternate score represents the degree to which the alternate allele affects the TF binding motif