This guide to the modules on the Gene page is illustrated in the Common Metabolic Diseases Knowledge Portal (CMDKP) but applies to all Knowledge Portals that display genetic and genomic results.
Click on a link to see documentation for that section of the Gene page:
Gene name and links to Region page
Tissue-specific gene expression
From the portal home page, you can navigate to the Gene page by entering a gene name into the search box and selecting Search gene. To navigate to a Gene page from a Region page, click one of the blue boxes near the top of the page to go to that Gene page:
You can also navigate to a Gene page by clicking on a gene name anywhere it appears on a Portal page.
Begin new search
At the top of the Gene page, the Begin new search box allows you to navigate to a different Gene, Variant, Region, Phenotype, or Tissue page. Begin typing in the box and select an option from the list.
Gene name and links to Region page
The Gene name section displays the primary gene name and offers links to navigate to the Region page around that gene:
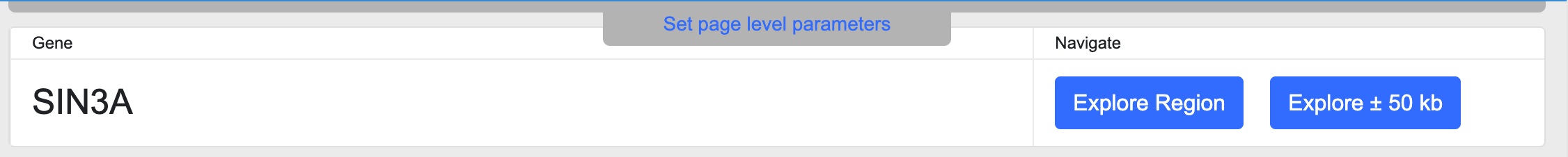
The Explore Region button takes you to a Region page whose coordinates are defined by the coding sequence of the gene, while the Explore +/- 50kb button takes you to a Region page spanning the coding sequence and 50kb of flanking sequences in either direction.
Functional associations
The Functional associations section of the Gene page displays information about the gene and its product, derived from other resources.

The Function tab shows a summary from UniProt, while the Gene Ontology and Pathways tabs display annotations served via MyGene.info. All information on the Function tab is retrieved dynamically.
Gene-level associations
In the next section, gene-level association scores generated by three different methods are represented on three tabs:
HuGE scores
HuGE scores, generated by the HuGE Calculator tool, incorporate both common-variant and rare-variant associations to quantify genetic support for involvement of a gene in a trait or disease. The HuGE score plot displays scores for the gene whose page you are viewing, across all phenotypes included in the portal you are viewing. To see all phenotypes in the Knowledge Portals, view a gene page in the Association to Function Knowledge Portal (A2FKP).

In the plot, the upper dotted line indicates Very strong evidence (HuGE score = 30) and the lower dotted line indicates Moderate evidence (HuGE score = 3). Mouse over a point to see the phenotype, the HuGE score, and the common- and rare-variant Bayes factors.
HuGE scores and Bayes factors are also listed in the table. Click the Open button to navigate to the interactive HuGE Calculator interface.
Common variant gene-level associations
The Common variant gene-level associations section of the Gene page displays gene-level associations for the gene across all phenotypes available in the Portal, and the associations are also listed in the table below.
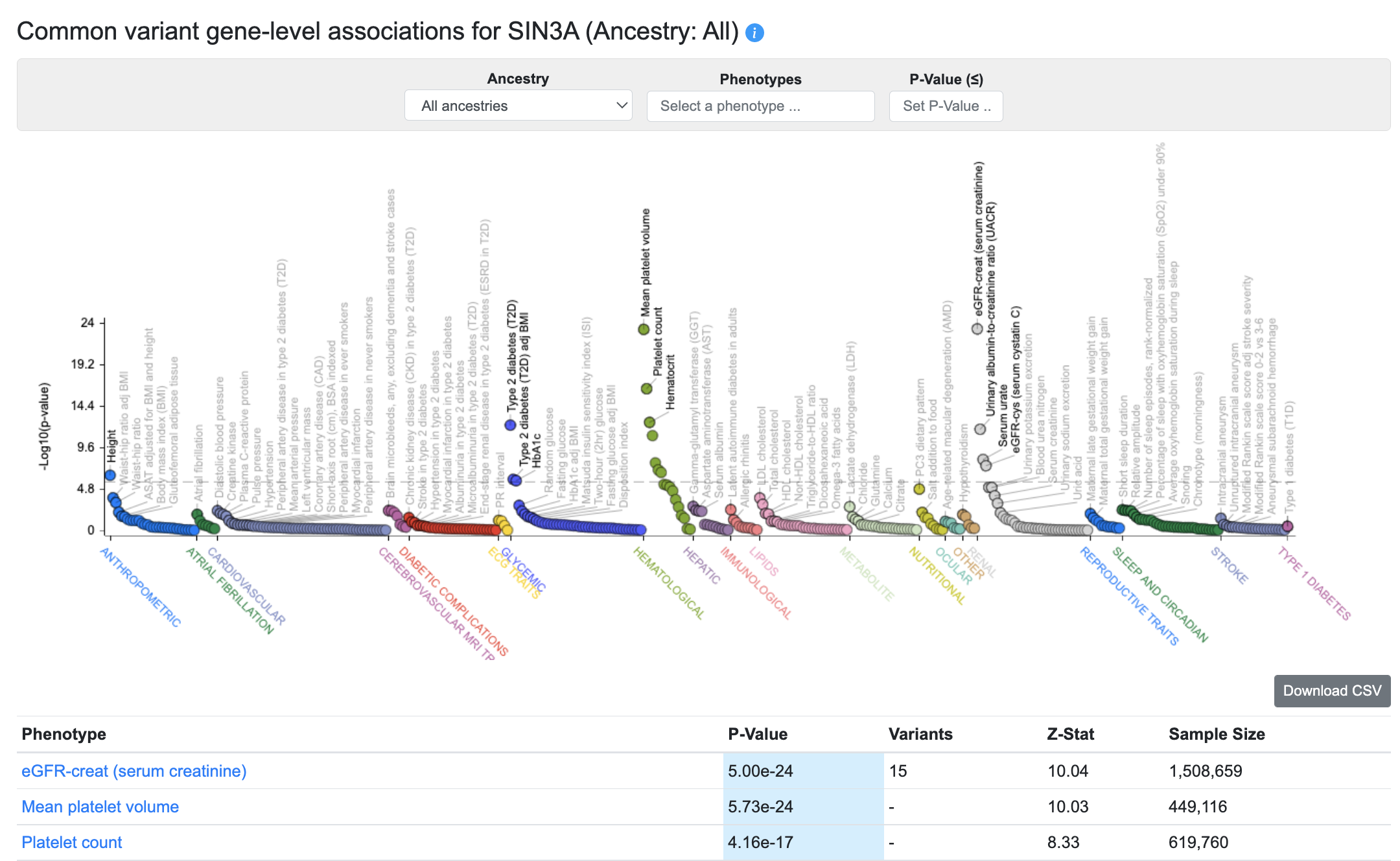
These gene-level associations are calculated from bottom-line common variant genetic associations using the MAGMA algorithm. A generally accepted threshold for significance of MAGMA results is p ≤ 2.5e-6. It should be remembered that although a MAGMA significant result indicates that the gene is close to a significantly associated variant, this does not necessarily mean that the gene itself is causal for the phenotype. If multiple genes are located near to a significantly associated variant, MAGMA assigns low p-values to all of them.
Using the menus above the plot, you can choose to view MAGMA associations from a single ancestry, or set a p-value threshold for the associations displayed on the plot and listed in the table. You can also view associations for a custom set of phenotypes, by entering phenotypes successively into the Phenotypes selection box.
Rare variant gene-level associations
The Rare variant gene-level associations plot and table display gene-level associations calculated from rare variants. The underlying data come from the following datasets:
- The AMP T2D-GENES T2D exome sequence analysis (T2D associations), published in Flannick et al. 2019. These associations were calculated for the gene overall (the canonical transcript) and for each individual transcript.
- The AMP T2D-GENES quantitative trait exome sequence analysis (23 cardiometabolic traits), published in Dornbos et al. 2022. These associations were calculated for the gene overall (the canonical transcript) and for each individual transcript.
- A rare coding variant analysis based on exome and whole-genome sequence data, from Jurgens, Wang, et al, with gene-level associations for 601 diseases across more than 750,000 individuals. On the Gene page, associations from this study are displayed only for phenotypes that map exactly to Knowledge Portal phenotypes. These associations were calculated for only the gene overall (the canonical transcript).
- Gene-level association scores derived from the Genebass resource and based on exome sequencing data from the UK Biobank. Associations from this resource are displayed for 189 phenotypes that map exactly to Knowledge Portal phenotypes. These associations were calculated for only the gene overall (the canonical transcript).
Results are displayed in the following order of priority:
- All results from the AMP T2D-GENES analyses are displayed
- Results from the Jurgens, Wang, et al. study are displayed for phenotypes that 1) map exactly to phenotypes included in the Knowledge Portal that you are viewing and 2) are not present in the AMP T2D-GENES results
- Results from Genebass are displayed for phenotypes that 1) map exactly to phenotypes included in the Knowledge Portal that you are viewing and 2) are not present in the Jurgens, Wang, et al. results or the AMP T2D-GENES results
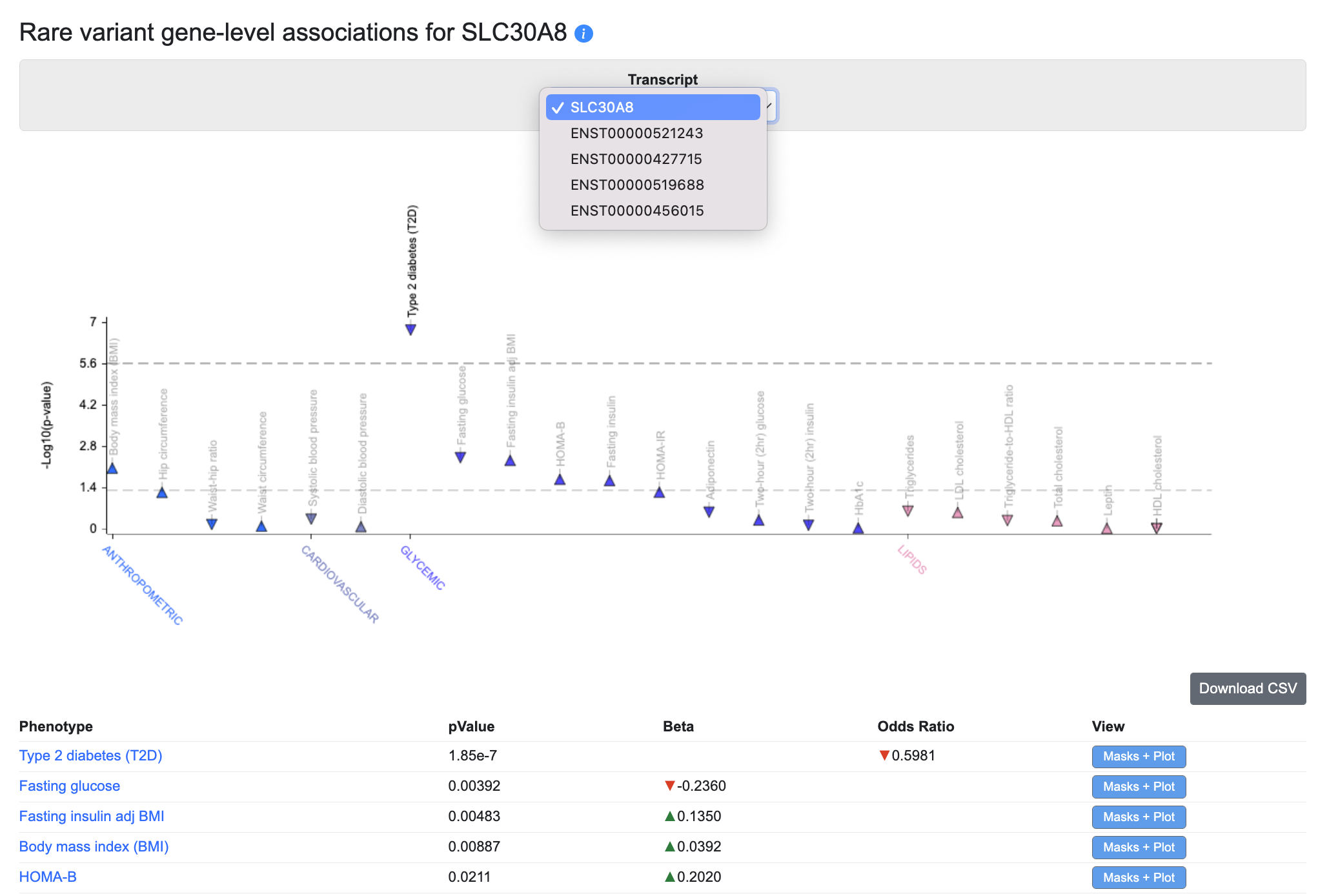
For each phenotype and gene, a burden test was performed to calculate a gene-level association score based on the associations of variants within the coding sequence. On the plot, the upper dotted line indicates exome-wide significance (p ≤ 6.57e-7), while the lower dotted line indicates nominal significance (p ≤ 0.05). Using the Transcripts menu, you can also choose to see gene-level associations for specific transcripts, where available. Tests were performed using different "masks" (criteria for grouping variants into sets by their predicted impact); the default view of the plot and table show the lowest p-value calculated across all the masks. See this page for more details about masks.
Click on the Masks + Plot button in the table to see the gene-level association scores for the variants retrieved by each mask, along with a forest plot showing their effect sizes and standard errors:
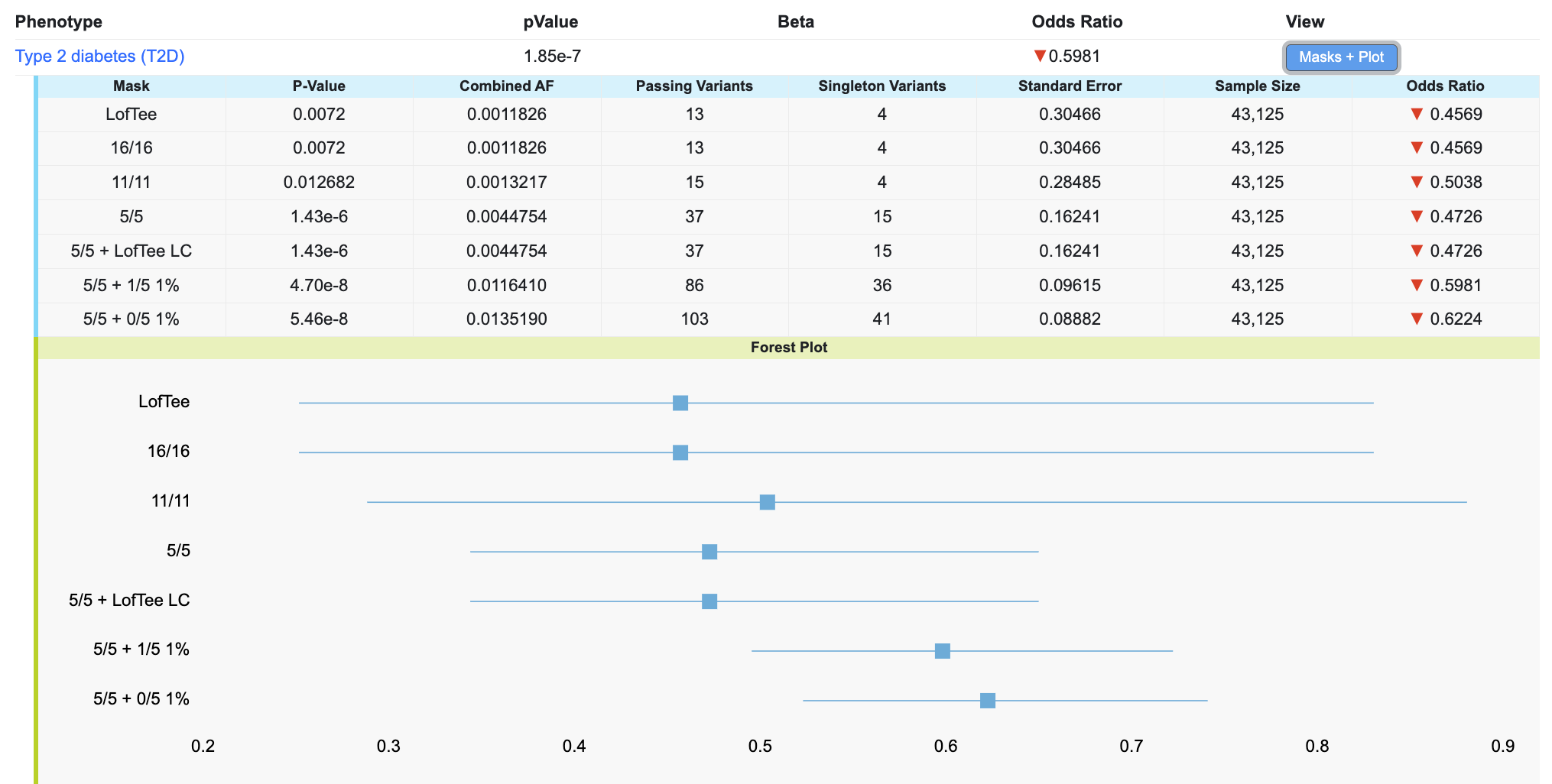
In the table, the Passing Variants column indicates the number of variants in the analysis, while the Singleton Variants column indicates the number of variants that were only observed in one person.
Colocalization results
This section displays results of colocalization of GWAS variants with eQTL variants, from the CoLocus browser.

Find extensive documentation and tutorials at the CoLocus site.
Tissue-specific gene expression
This section of the Gene page displays tissue-specific gene expression levels in a graphic and table. Currently, bulk tissue expression levels are shown; in the future, we plan to include single-cell gene expression levels. To see the provenance of the data and view the original datasets in the Common Metabolic Diseases Genome Atlas, click the Show Datasets button in the table.
The menus above the table allow you to:
- View the graph in linear or logarithmic scale
- Filter the datasets shown in the plot and table by minimum sample count
- Filter the datasets shown in the plot and table by their source
- Display expression levels in disease states, where available
In the graphic, each tissue column represents multiple datasets from tissues within that category. When you mouse over a specific point, all other points originating from that dataset are highlighted.
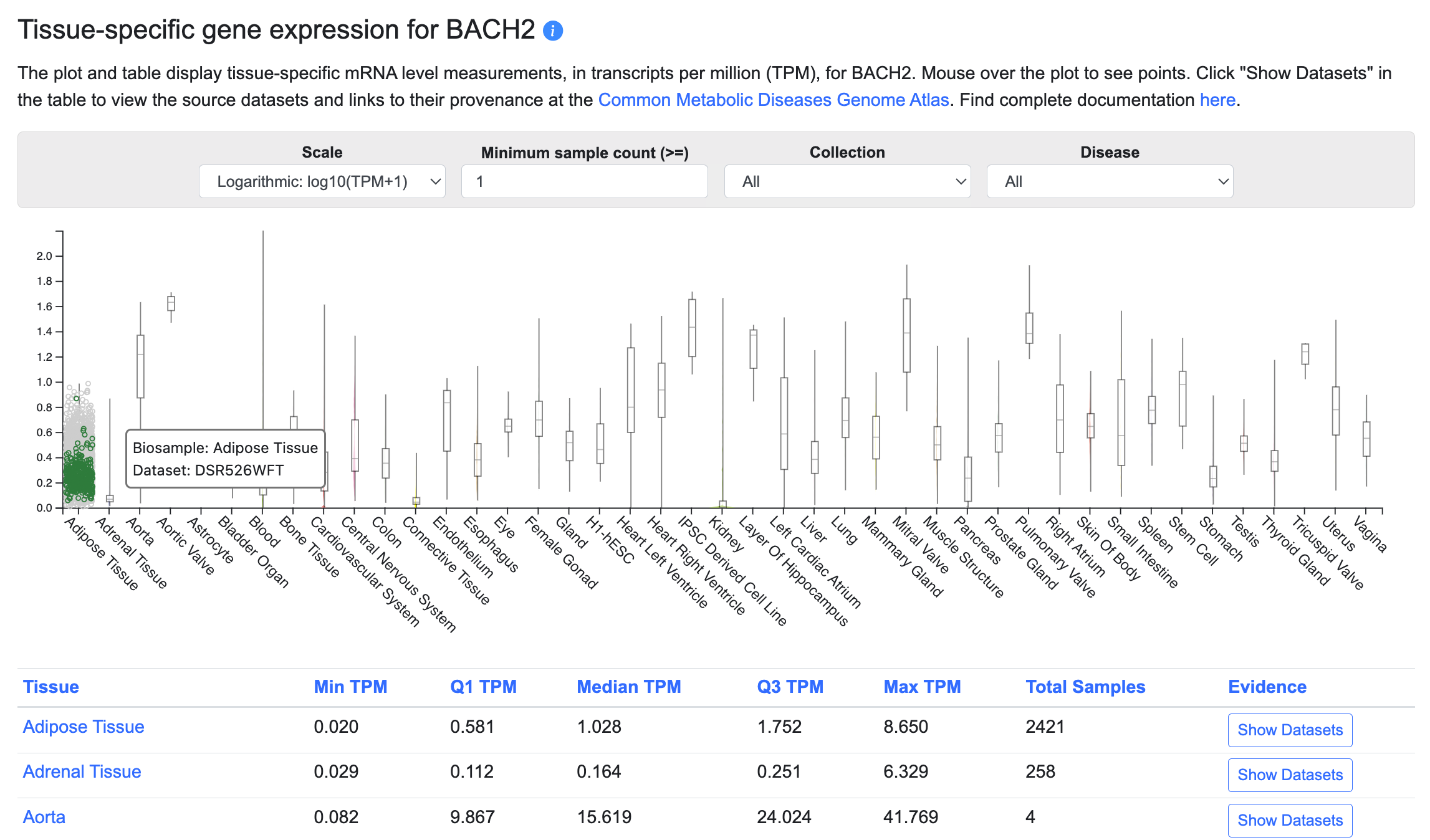
In the table, click on any column header to sort by that column. Click the Show Datasets button to see details about the individual datasets grouped within each tissue category, including their specific tissue or cell type (the Biosample), transcripts per million (TPM) values, sample sizes, and links to the original data at CMDGA.
Predicted effector gene lists
Many genetic association studies now include predictions of the most likely effector (causal) gene at each GWAS locus, usually generated by considering multiple types of evidence. You may browse and filter these lists on our Predicted Effector Genes page. When viewing a gene page, if that gene is predicted to be an effector gene, the predicted effector gene lists on which it appears will appear in a table:
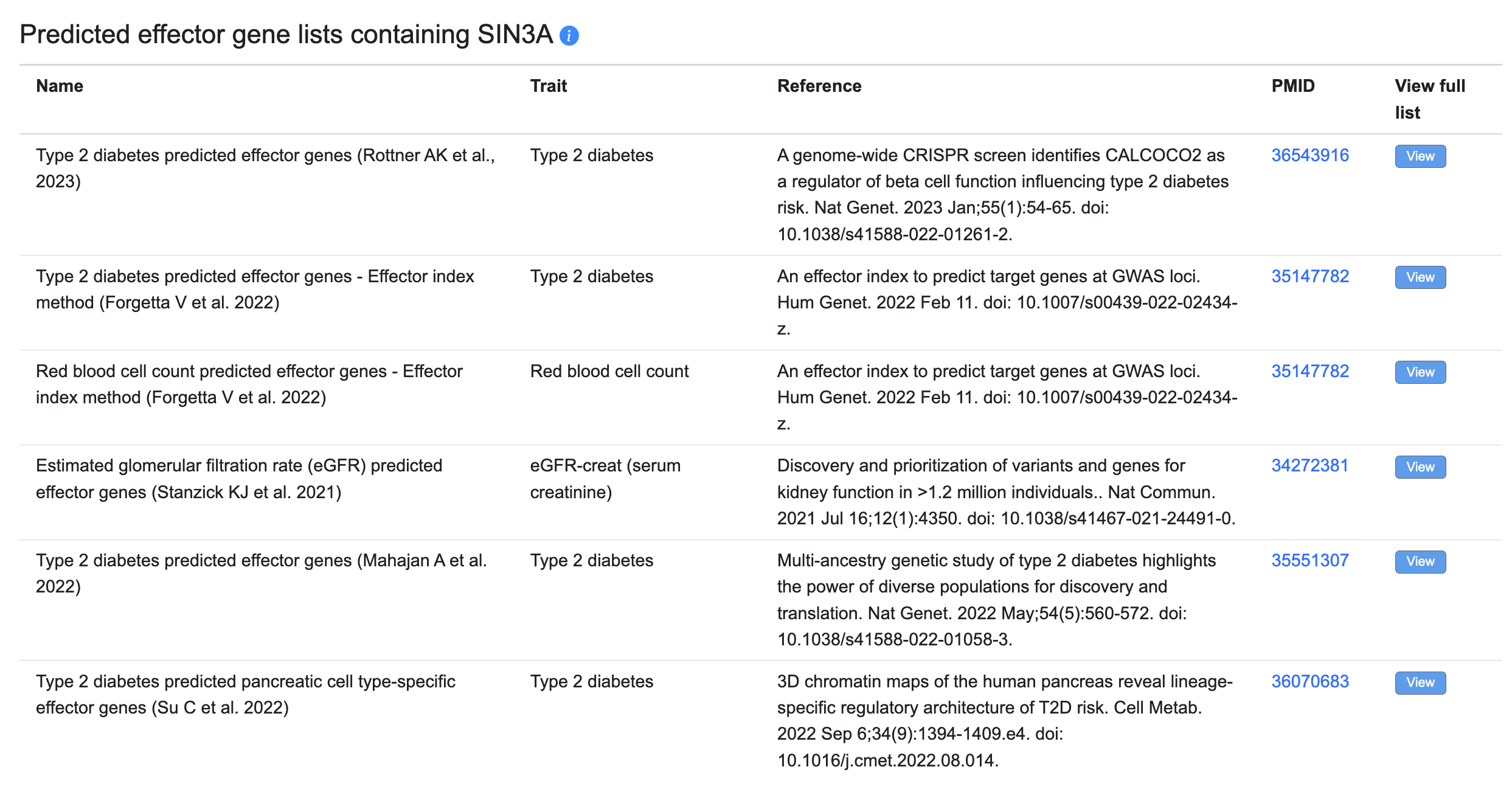
The table displays the name of each list; the trait for which it predicts effector genes; and the reference and PubMed ID (linked to its PubMed record). The View buttons lead to each effector prediction list.
UniProt cross-references
The UniProt cross-references section of the Gene page displays accession numbers for records of the gene, its transcript, and its protein product, derived from UniProt.
External resources
The External resources section of the gene page displays links to records for the gene in other resources: Ensembl, HUGO Gene Nomenclature Committee (HGNC), Mouse Genome Informatics (MGD), Rat Genome Database (RGD)