The Genetic Association Interactive Tool (GAIT) allows you to access rare variant associations to test whether a gene of interest is genetically associated with a phenotype of interest.
Rare variant associations can be especially informative because they often have much larger effects than common variants. But because these variants are rare, single-variant genetic associations don’t have much statistical power. Instead, gene-level associations calculated from the aggregate burden of rare variants in a gene can suggest correlations of genes with diseases and traits.
GAIT performs gene-level association analysis using custom sets of variants. It can be used as part of a reverse genetics approach, where a researcher starts with a gene of interest and evaluates the effect of perturbing that gene on traits of interest. Our rare-variant gene-level associations workflow illustrates a scientific use case for GAIT.
GAIT can be most useful for researchers who have already studied a gene in detail and know which variants might be the most damaging - for example, those located in a domain known to be important for function, or those shown to affect gene product function in an in vitro assay. In this case, the "masks" (variant filters) can be used to define a set of potentially impactful variants, which can then be used to test whether these variants, in aggregate, have a significant association with a phenotype of interest.
The starting point for GAIT is a specific gene of interest. Use GAIT to see whether variants in the gene or transcript have an aggregate effect on a phenotype of interest.
The figure below illustrates the overall GAIT workflow:
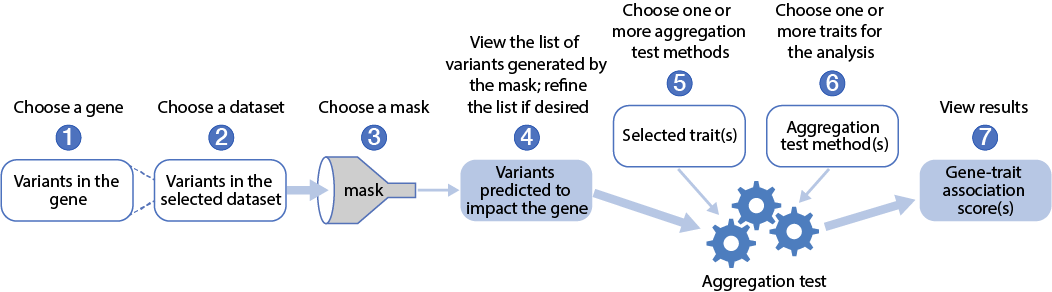
- Choose a gene
- Choose a dataset
- Choose a mask
- View the list of variants generated by the mask; refine the list if desired
- Choose one or more traits for the analysis
- Choose one or more aggregation test methods
- View results
In brief, with each step you will successively define the set of variants to analyze, and then you will run an aggregation test using those variants to generate a gene-level score for association of your gene with a phenotype of interest.
The video below gives a high-level overview of the tool.
Step 1. Choose a gene.

To perform custom aggregation analysis using GAIT, begin typing the name of your gene of interest, and select the correct name:
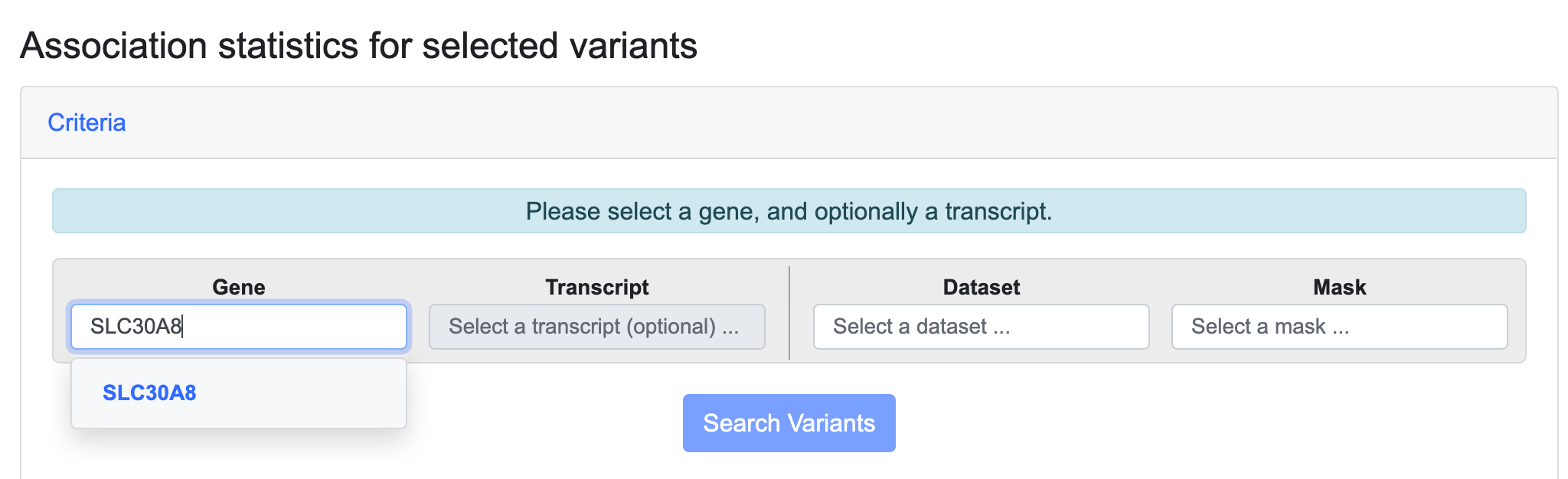
After selecting a gene, you may optionally select a specific transcript of the gene:
![]()
If you don't select a specific transcript, the analysis is done using the coordinates of the Ensembl canonical transcript.
Step 2. Choose a dataset.
Choose a dataset that includes the phenotype(s) for which you want to test your gene's association.
- "52K" indicates whole-exome sequence data generated by Flannick et al. 2019 and Dornbos et al. 2022. Choose this dataset to do association analysis for: type 2 diabetes; fasting glucose; fasting insulin; two-hour glucose; two-hour insulin; diastolic blood pressure; systolic blood pressure; HDL cholesterol; LDL cholesterol; total cholesterol; triglycerides; or body mass index (BMI).
- "TOPMed" refers to the exome subset of whole-genome sequence generated by the TOPMed consortium (Wessel et al. 2020; DiCorpo et al. 2022). Choose this dataset to do association analysis for type 2 diabetes, fasting glucose, or fasting insulin.
How does it work? GAIT securely accesses individual-level genetic association data from these datasets to perform aggregation analysis, using LDserver to calculate score statistics and covariance matrices from the individual genotypes and phenotypes. LDserver responds to a REST query (specifying a list of variants) from GAIT and either computes these statistics on-demand (for the 52K dataset) or accesses pre-computed files of genome-wide statistics (for the TOPMed dataset). It then converts these statistics to an aggregate association statistic. Individual-level data stored within LDserver are never directly accessible via the Knowledge Portal.
Select a dataset as shown below:
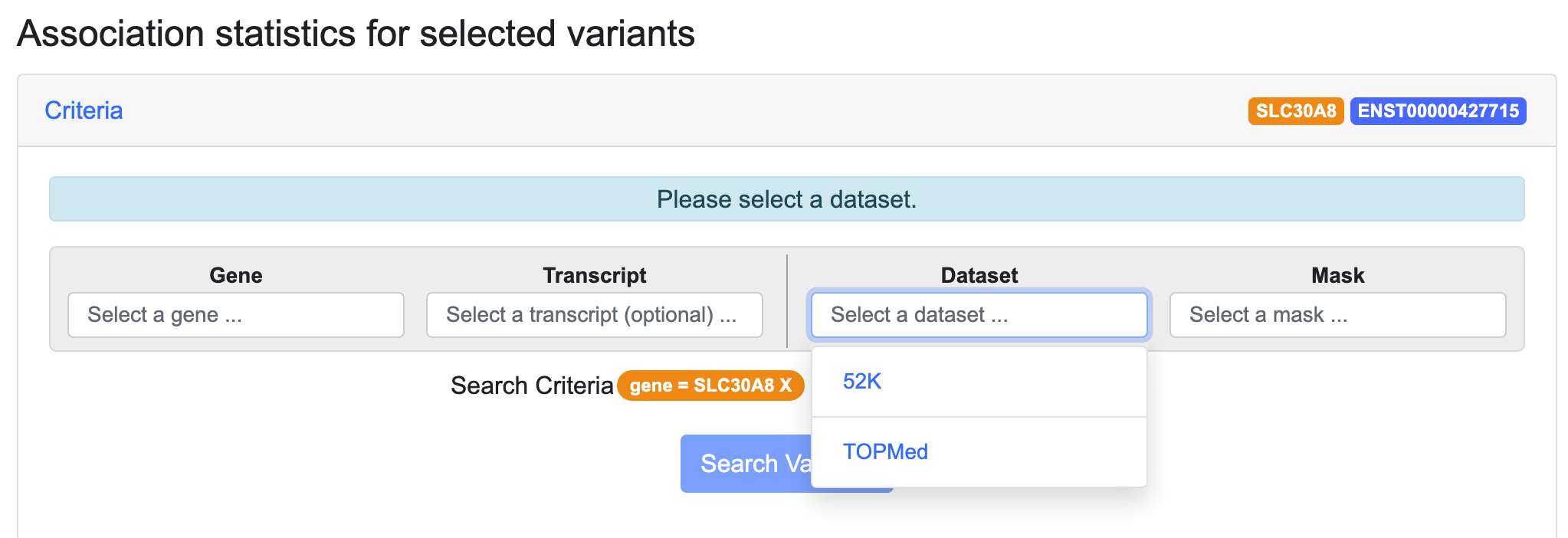
Once you have selected a dataset, GAIT retrieves all of the variants in that dataset that are within your chosen gene.

Step 3. Choose a mask.
A mask is a set of criteria that you can use to filter the set of variants in order to do the aggregation test with the variants that are most likely to affect the gene product, eliminating the variants that have little or no effect. The masks filter the variants in your gene of interest by their categories or scores generated by multiple tools that are commonly used to estimate the deleteriousness of variants. The masks are listed on the menu in order of stringency, from most to least; i.e., LofTee is the most stringent mask and will retrieve the smallest number of variants that have the most severe predicted impact.
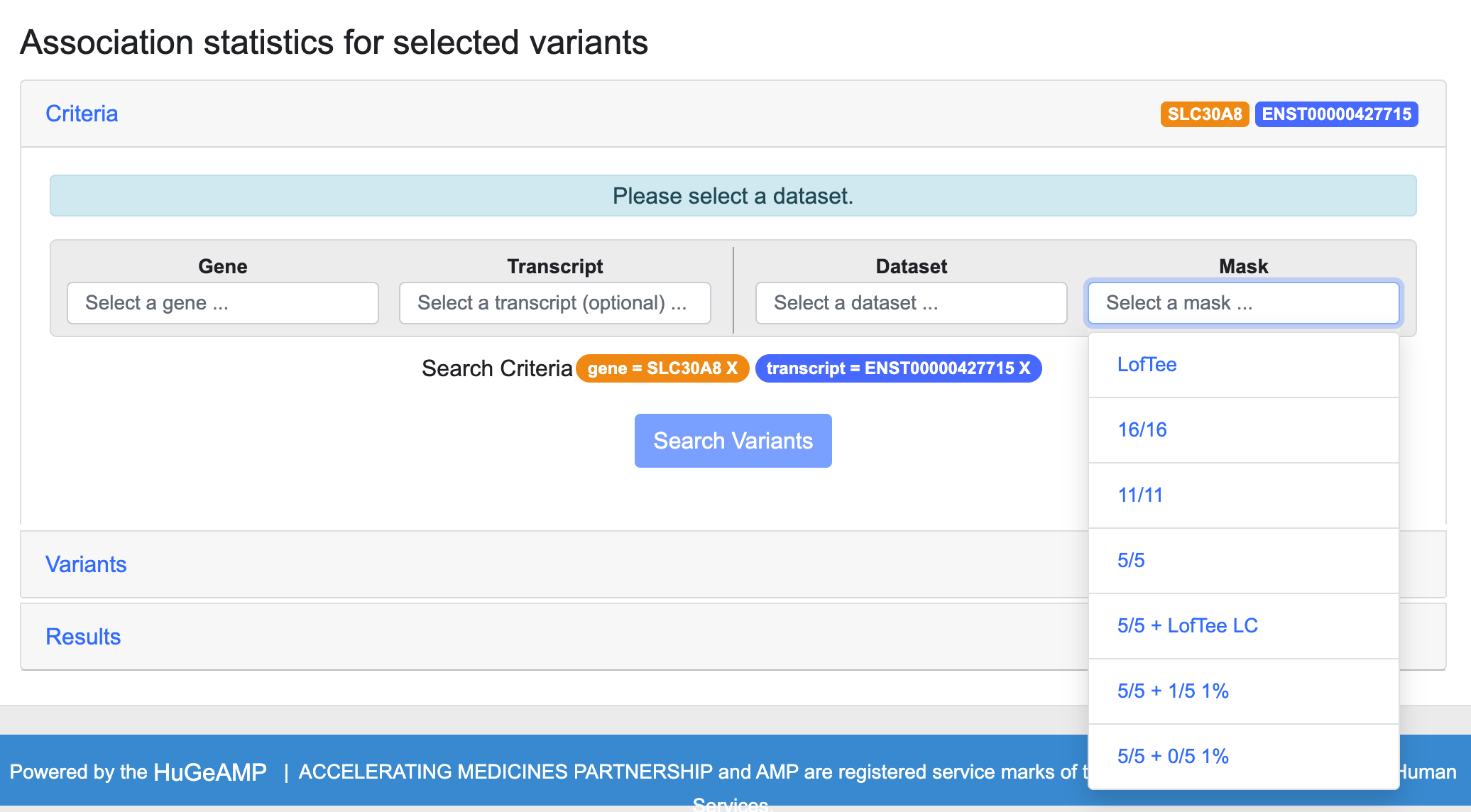
The tools used to predict variant deleteriousness and their categories or thresholds are:
- LofTee HC: Variants predicted with high confidence by the Loss-Of-Function Transcript Effect Estimator (LofTee; Karczewski et al, 2020 ) to cause a loss of function of the encoded protein
- VEST3>90%: Variants predicted with > 90% probability to be deleterious by the Variant Effect Scoring Tool VEST 3.0 (Carter et al., 2013)
- CADD>90%: Variants predicted with > 90% probability to be deleterious by the Combined Annotation Dependent Depletion tool CADD (Rentzsch et al., 2019)
- DANN>90%: Variants predicted with > 90% probability to be deleterious by the Deleterious Annotation of genetic variants using Neural Networks DANN tool (Quang et al., 2015)
- Eigen-raw>90%: Variants predicted with > 90% probability to be deleterious by the Eigen method (Ionita-Laza et al., 2016)
- Eigen-PC-raw>90%: Variants predicted with > 90% probability to be deleterious by the Eigen-PC method (Ionita-Laza et al., 2016)
- FATHMM pred=D: Variants predicted to be deleterious by the Functional Analysis through Hidden Markov Models (FATHMM) method (Shihab et al., 2013).
- FATHMM-MKL pred=D: Variants predicted to be deleterious by the Functional Analysis through Hidden Markov Models (FATHMM-MKL) method (Shihab et al., 2013).
- PROVEAN pred=D: Variants predicted to be deleterious by the Protein Variation Effect Analyzer (PROVEAN) method (Choi and Chan, 2015).
- MetaSVM pred=D: Variants predicted to be deleterious by the MetaSVM method (Dong et al., 2015)
- MetaLR pred=D: Variants predicted to be deleterious by the MetaLR method (Dong et al., 2015)
- MCAP>0.025: Variants with a Mendelian Clinically Applicable Pathogenicity (M-CAP) (Jagadeesh et al., 2016) score over 0.025.
- PolyPhen HDIV pred=D: Variants predicted to be deleterious by the PolyPhen HDIV method (Adzhubei et al., 2010)
- PolyPhen HVAR pred=D: Variants predicted to be deleterious by the PolyPhen HVAR method (Adzhubei et al., 2010)
- SIFT pred=del: Variants predicted to be deleterious by the Sorting Intolerant From Tolerant (SIFT) algorithm (Kumar et al, 2009 )
- LRT pred=D: Variants predicted to be deleterious by the Likelihood Ratio Test (LRT; Chun and Fay, 2009).
- MutTaster pred=(D or A): Variants predicted to be probably deleterious (D) or variants known to be deleterious (A) by the MutationTaster method (Schwarz et al., 2010)
- VEP impact=HIGH: Variants predicted to have high impact by the Ensembl Variant Effect Predictor (VEP)
- VEP impact=MOD: Variants predicted to have moderate impact by the Ensembl Variant Effect Predictor (VEP)
- LofTee LC: Variants predicted with low confidence by the Loss-Of-Function Transcript Effect Estimator (LofTee; Karczewski et al, 2020 ) to cause a loss of function of the encoded protein
- Max MAF<1%: Variants with minor allele frequency less than 1%
In the table below, each column indicates a mask, and an "X" indicates that variants meeting the criterion in that row are included in the mask. For example, the mask named "16/16" includes all variants within the selected gene that are predicted with high confidence by LofTee to cause loss of function plus those that have scores of > 90% from any of the VEST3, CADD, DANN, Eigen-raw, and Eigen-PD-raw methods.
| Mask | |||||||
|---|---|---|---|---|---|---|---|
| Criterion | LofTee | 16/16 | 11/11 | 5/5 | 5/5 + LofTee LC 1% | 5/5 + 1/5 1% | 5/5 + 0/5 1% |
| LofTee HC | x | x | x | x | x | x | x |
| VEST3>90% | x | x | x | x | x | x | |
| CADD>90% | x | x | x | x | x | x | |
| DANN>90% | x | x | x | x | x | x | |
| Eigen-raw>90% | x | x | x | x | x | x | |
| Eigen-PC-raw>90% | x | x | x | x | x | x | |
| FATHMM pred=D | x | x | x | x | x | ||
| FATHMM-MKL pred=D | x | x | x | x | x | ||
| PROVEAN pred=D | x | x | x | x | x | ||
| MetaSVM pred=D | x | x | x | x | x | ||
| MetaLR pred=D | x | x | x | x | x | ||
| MCAP>0.025 | x | x | x | x | x | ||
| PolyPhen HDIV pred=D | x | x | x | x | |||
| PolyPhen HVAR pred=D | x | x | x | x | |||
| SIFT pred=del | x | x | x | x | |||
| LRT pred=D | x | x | x | x | |||
| MutTaster pred=(D or A) | x | x | x | x | |||
| VEPimpact=HIGH | x | ||||||
| VEPimpact=MOD | x | x | |||||
| LofTee LC | x | ||||||
| Max MAF<1% | x | x | x | ||||
Which mask to choose? Choosing a mask is a key step in GAIT, because the association test will only produce meaningful results if it is run using a set of variants of which most affect the function of the gene product. The number of variants retrieved by each mask will vary widely between genes, which is the reason that GAIT offers so many masks.
In general, it may be most informative to start with the most stringent mask (LofTee: variants likely to cause loss of function) and to ease the stringency if few variants are retrieved. Since GAIT runs quickly, you can change the masks in multiple tests to explore how association scores change with different masks.
After selecting a mask, click the "Search variants" button. You will be presented with the list of variants in the selected gene, transcript, and dataset that meet the mask criteria.


To add columns to the table showing variant scores from individual deleteriousness prediction methods, check the boxes above the table. You may select or deselect individual variants to refine the set of variants to use for the aggregation test.
Should I refine the list of variants by selecting or eliminating individual variants?
If you have prior knowledge about the gene and/or variants within it, it's a good idea to refine the list. There are two major use cases for refining the list:
1. You know something about the structure of the gene and which domains or residues are important. For example, when querying for a transcription factor you could refine the variant list to include only missense or nonsense mutations within the DNA-binding domain.
2. You want to dissect an association signal in order to determine the contribution of specific variants. In this case, you can run GAIT while including and excluding specific variants to see how the association score changes. An example of this usage is outlined in our rare variant gene-level associations workflow.
Step 5. Choose one or more traits for the analysis.

Next, select one or more phenotypes for the analysis from the Phenotypes menu above the variant table. The range of phenotypes available is determined by which dataset you chose in step 2 (see above).
Step 6. Choose one or more aggregation test methods.

The available aggregation test methods are Collapsing Burden, Variable Threshold, SKAT, and SKAT-Optimal (SKAT-O). Since GAIT allows you to choose multiple methods, we encourage you to choose all of them and compare the results. Some considerations for using each method:
- The Collapsing Burden method (Goldstein et al. 2013) assumes that all variants in the set have the same effect on the gene. The results would be most accurate when using a small set of variants that are likely damaging, as would be obtained by using a stringent mask such as LofTee.
- The SKAT method (Sequence Kernel Association Test; Wu et al. 2011) assumes that only a few variants in a set are impactful and that the set includes variants that are benign or even have the opposite direction of effect. This method is better for larger sets of variants obtained by using lenient masks. Note that the results from SKAT do not include direction of effect.
- The SKAT-O method (SKAT-Optimal; Lee, Wu, and Lin 2012) tries to strike a balance between the assumptions of the collapsing burden and SKAT methods, and may be the best choice for new GAIT users. Note that the results from SKAT-O do not include direction of effect.
- The Variable Threshold method (Price et al. 2010) is a modification of the collapsing burden test. It determines the optimal frequency threshold at which to exclude common variants, and dynamically excludes them. This method could be appropriate when your set of variants includes common variants that may not be benign.
Step 7. View results.

After making all selections, click the Run Analysis button. Results are presented in a table for each phenotype.

See the Rare variant gene-level associations workflow for an example of how to ask scientific questions using GAIT.