This guide to the modules on the Phenotype page is illustrated in the Common Metabolic Diseases Knowledge Portal (CMDKP) but applies to all Knowledge Portals that display genetic and genomic results.
In the Knowledge Portals we curate phenotypes from GWAS publications based on the traits reported by the authors. We categorize phenotypes into "Phenotype groups" (e.g., "CARDIOVASCULAR", "RENAL", "GLYCEMIC"), which may change over time. See this page for an overview of the phenotypes available in each portal and the phenotype groups to which they belong. We are currently mapping portal phenotypes to Experimental Factor Ontology (EFO) terms to facilitate searching and coordination with the GWAS Catalog.
Each Knowledge Portal displays genetic associations for phenotypes within a defined set of phenotype groups that represent the phenotypes most relevant to the disease focus of that portal. For example, the Type 2 Diabetes Knowledge Portal includes phenotypes in the GLYCEMIC group but not those in the NEUROLOGICAL group. Try the new Association to Function Knowledge Portal to view associations for all phenotypes or set your own custom phenotype view.
Click on a link to see documentation for that section of the Phenotype page:
Top single-variant associations
You can navigate to the Phenotype page by clicking the Phenotype tab on the portal home page, starting to type a phenotype, and then selecting the phenotype from the suggestions that appear. You can also go to a Phenotype page by clicking on a phenotype name anywhere it appears on the portal.
New search/set ancestry
The top of the Phenotype page offers two options:
- Begin new search: Begin typing a gene name, variant ID, phenotype, or tissue name and select an option from the menu to navigate to a different Knowledge Portal page.
- Ancestry: Click the menu to select an ancestry and change the Phenotype page to display only associations from that ancestry. Note that associations are not available for every ancestry-phenotype combination.
Page description
The page description section summarizes the content of the page and links to workflows and documentation:
Manhattan and Q-Q plots
The next section of the page displays a Manhattan plot of associations across the genome for that phenotype, and a quantile-quantile probability (Q-Q) plot giving an indication of the quality of the associations:
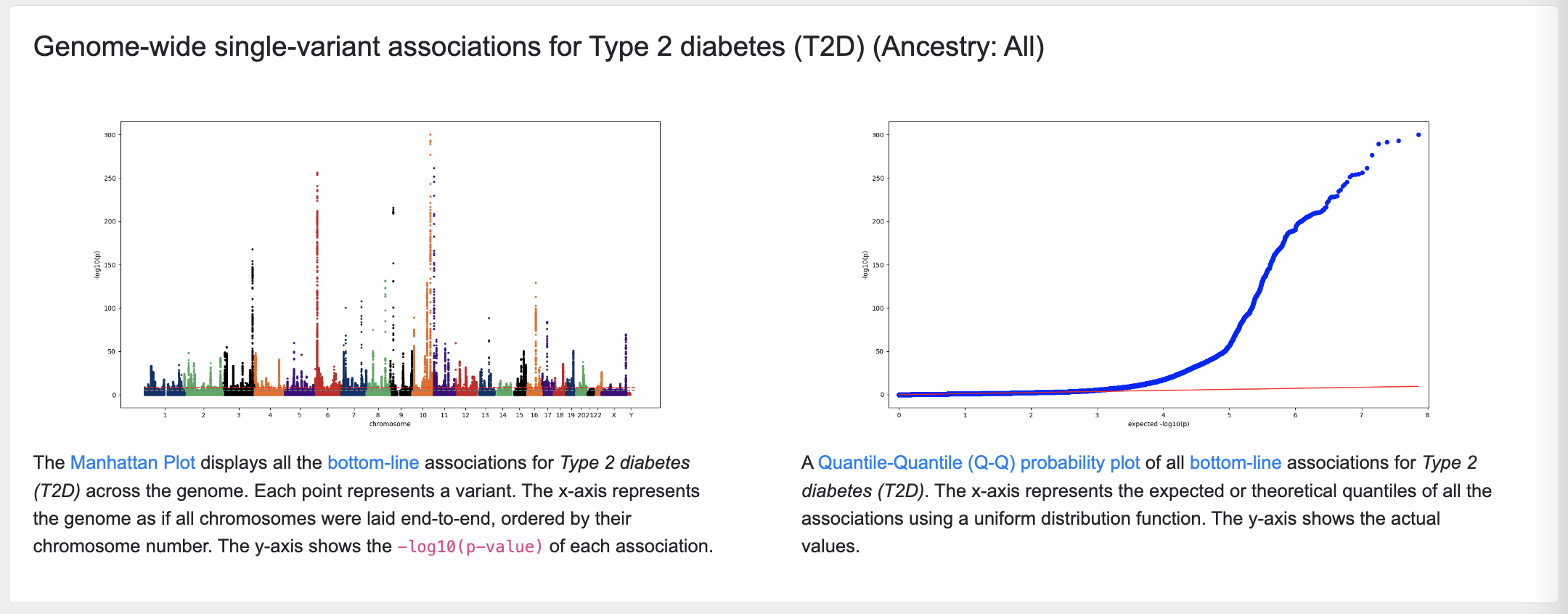
The associations shown on the page are derived from "bottom-line" integrative analysis.
Top single-variant associations
Below the plots, a table lists all bottom-line associations for the phenotype with p-value ≤ 5e-8:
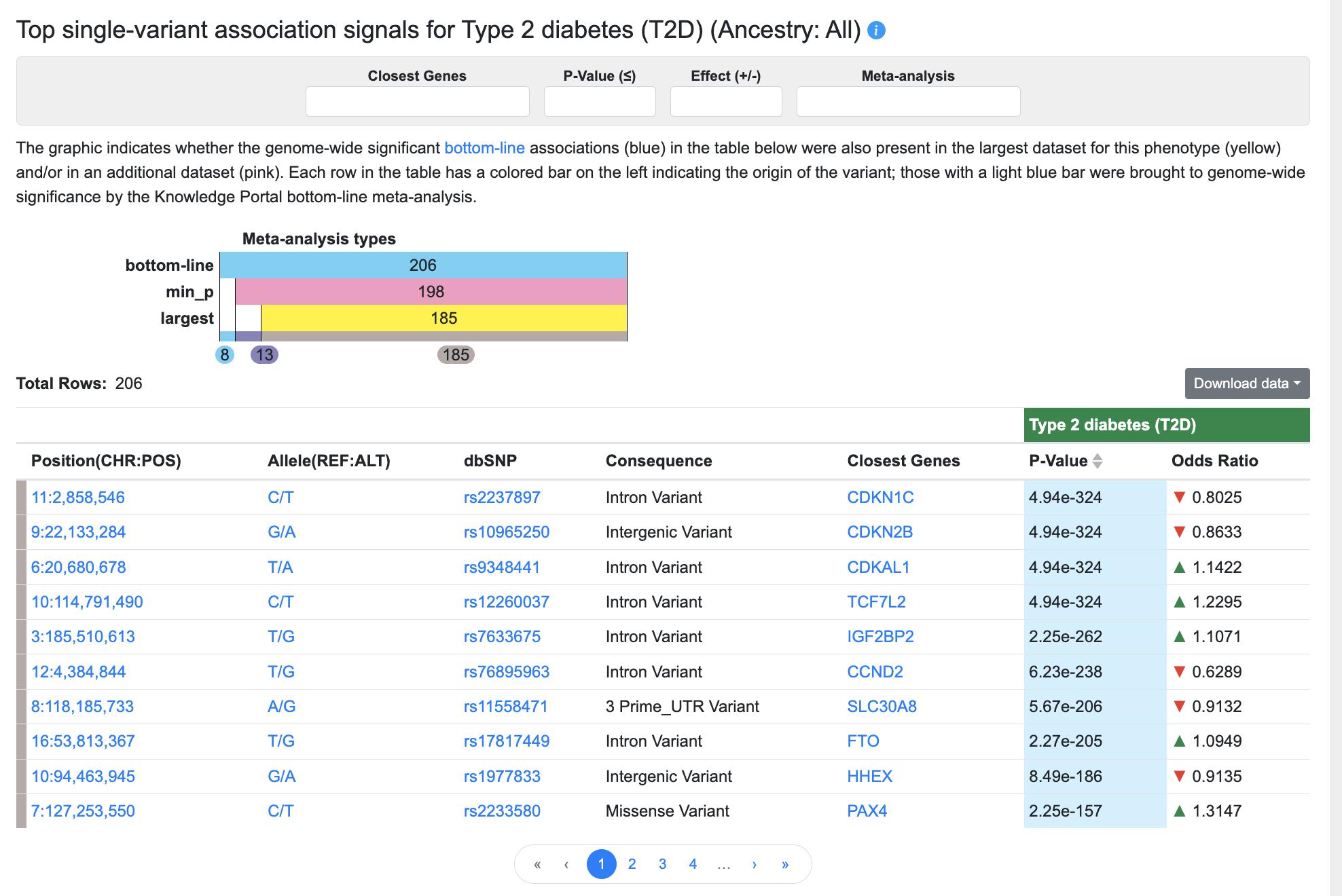
The graphic indicates whether the genome-wide significant (p ≤ 5 x 10e-8) bottom-line associations (blue) in the table below were also present in the largest dataset for this phenotype (yellow) and/or in an additional dataset (pink). Each row in the table has a colored bar on the left indicating the origin of the variant; those with a light blue bar were brought to genome-wide significance by the Knowledge Portal bottom-line meta-analysis.
You can filter the associations displayed in the table using the filters above the table:
- Begin typing in the Closest Genes box to select a gene in the region and filter the table to display the variants for which that gene is the closest (as determined by VEP).
- Enter a p-value in the format "5e-8" to set an upper threshold for the p-values of the associations displayed.
- Enter "positive" or "negative" into the Effect box to filter the table by association direction of effect.
- Make a selection from the Meta-analysis filter:
- Choose Bottom-line to display only associations that were brought to genome-wide significance by the bottom-line analysis
- Choose Bottom-line + Min_p to display associations that were present in the results of the bottom-line analysis AND were also present in an individual dataset (not the largest dataset for the phenotype)
- Choose Bottom-line + Min_p + Largest to display associations that were present in the results of the bottom-line analysis AND were also present in the largest individual dataset for the phenotype
The columns of the table are as follows:
- Position indicates the chromosome and coordinate of the variant, and is linked to a Region page centered on the variant and spanning 100kb.
- Allele shows reference allele/alternate allele and is linked to the Variant page.
- dbSNP shows the reference SNP number (rsID) assigned to this variant by dbSNP and is linked to the Variant page.
- Consequence indicates the predicted impact of the variant, as predicted by VEP.
- Closest genes indicates the one or more genes nearest to the variant, as predicted by VEP. Gene names are linked to their Gene pages.
- p-value represents the probability that the observed frequency difference would occur by chance. p-values in this table are bottom-line p-values derived from integrative analysis.
- Odds ratio (OR), reported for dichotomous (binary) traits, indicates the magnitude of the variant effect. OR greater than 1 indicates a positive correlation between the effect allele and a phenotype (green, upward-pointing arrows), while OR less than 1 indicates a negative correlation (red, downward-pointing arrows).
- Beta, reported for continuous (quantitative) traits, is the estimated difference in phenotype between a heterozygous carrier of an effect allele and a homozygous reference allele carrier. A beta coefficient greater than 0 indicates a positive correlation between the effect allele and a phenotype (green, upward-pointing arrows), while beta less than 0 indicates a negative correlation (red, downward-pointing arrows).
Click the Download CSV button to download the contents of the table in comma-separated values format.
Credible Sets to Cell Type
Results from the Credible Sets to Cell Type (CS2CT) method, developed by the Knowledge Portal team at the Broad Institute, can provide clues about the tissues that mediate the effects of causal variants. The method starts with credible sets–both curated from the literature and calculated from meta-analyzed bottom-line associations. The genomic locations of the credible sets are compared with the locations of tissue-specific epigenomic annotations to calculate their overlap. Each credible set is assigned an overlap probability, indicating the likelihood that the causal variant is located within the annotated region, and a “genericity” value that is a measure of the tissue specificity of the overlap.
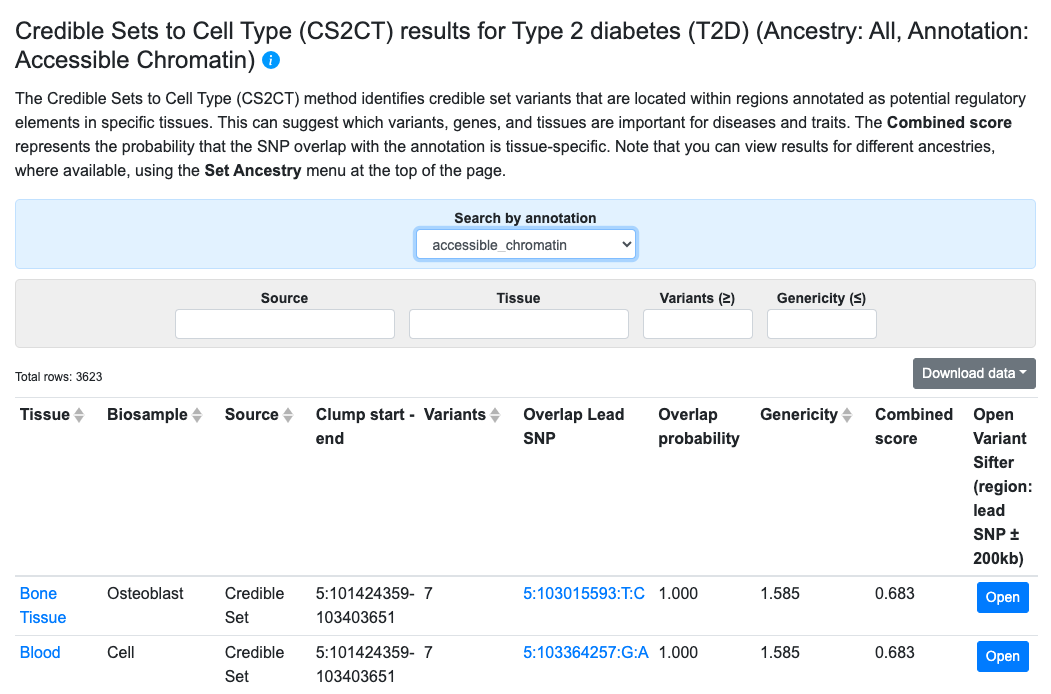
CS2CT results are displayed in a table, as shown above. The results shown by default are those for the annotation and tissue with the highest combined score. Metrics for the overlaps are expressed as:
- Overlap probability (higher value = greater probability of overlap)
- Genericity (lower value = greater tissue specificity of the overlap)
- Combined score (higher value = greater probability that the SNP overlaps the annotation in only one or a small number of tissues)
In the table, click the Open button to navigate to the Variant Sifter in the region of a credible set.
Filters above the table allow you to view results for different annotation types, credible set sources, or tissues, and to set a minimum threshold for number of variants in the credible set and a maximum threshold for genericity.
Datasets
This table lists the individual datasets whose results were included in the bottom-line integrative analysis:
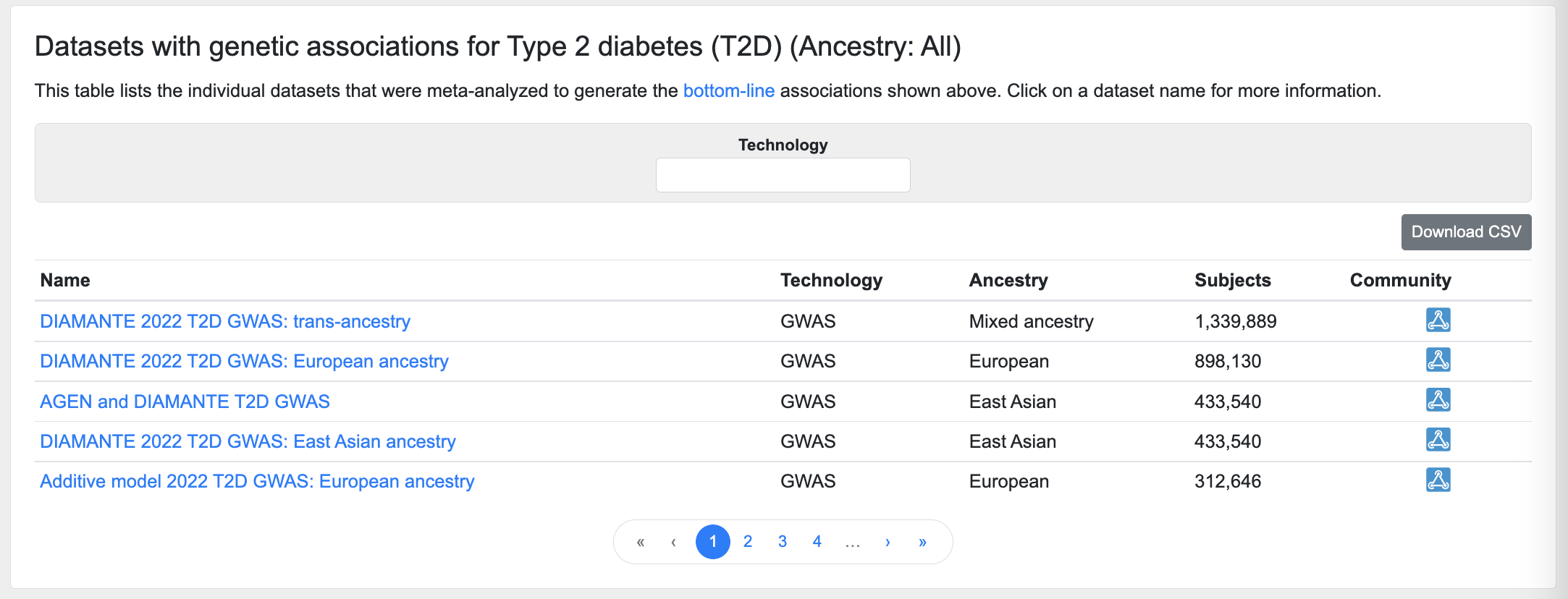
The table may be filtered by the technology used in the association study (e.g., GWAS, exome chip, etc.)
Click on a dataset name to see its Dataset Inspector page, which provides Manhattan and Q-Q plots and a table of top associations for that dataset, as well as the associated publication and information on downloading the summary statistics (if applicable).
An icon in the Community column indicates the disease-focused community that contributed the dataset to the Knowledge Portals.
Top gene-level associations
This section of the Phenotype page displays the top gene-level associations for the phenotype from three sources, on separate tabs:
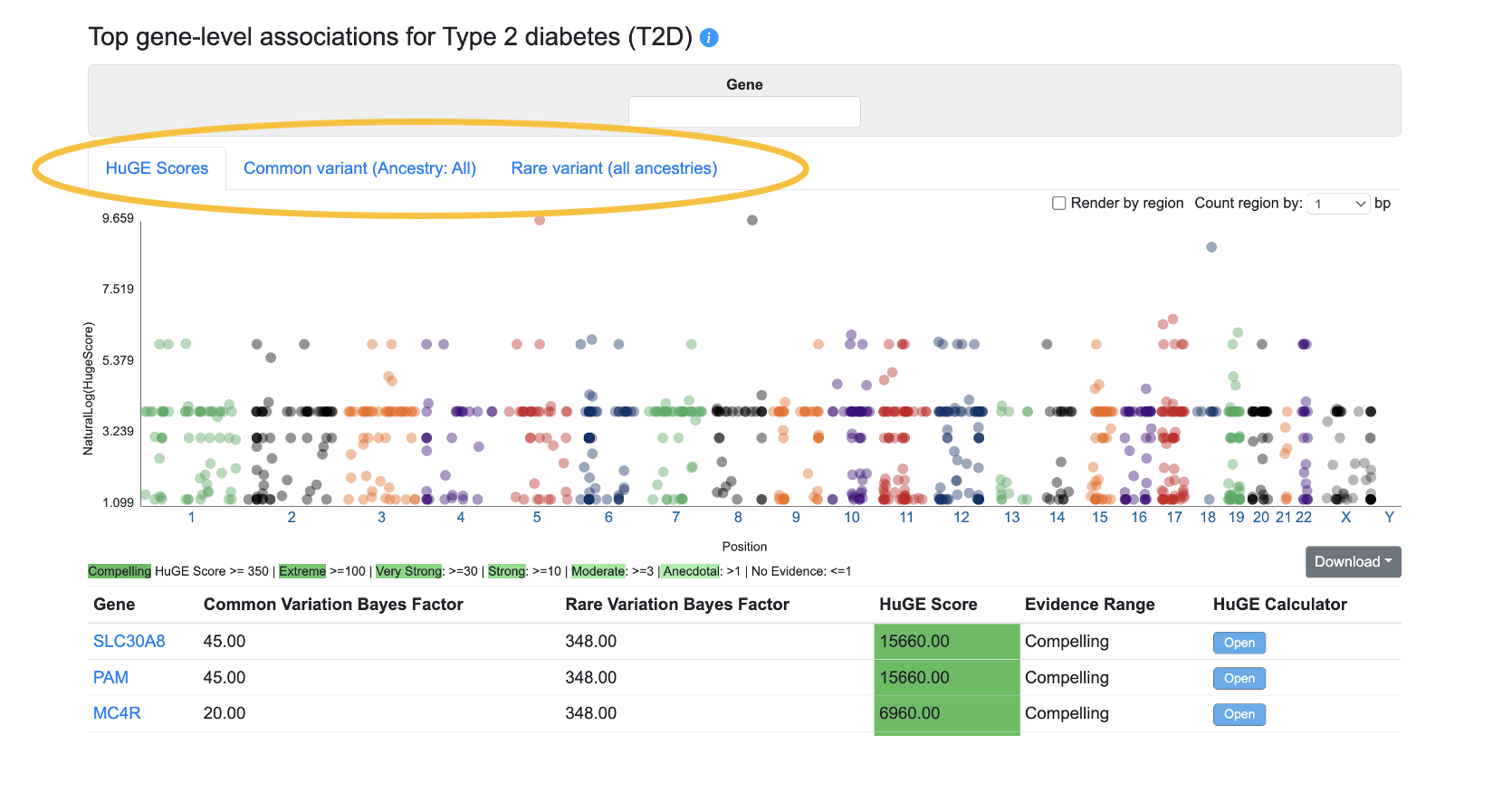
The HuGE Scores tab, shown by default, displays the genes with the highest HuGE Calculator scores for the phenotype whose page you are viewing. The HuGE Calculator uses both common-variant and rare-variant associations to weigh the evidence supporting involvement of a gene in a trait or disease, generating both a numerical score and a categorization of the evidence, such as "Compelling".
The Common variant tab displays gene-level associations calculated from bottom-line common variant genetic associations using the MAGMA algorithm. A generally accepted threshold for significance of MAGMA results is p ≤ 2.5e-6. It should be remembered that although a MAGMA significant result indicates that the gene is close to a significantly associated variant, this does not necessarily mean that the gene itself is causal for the phenotype. If multiple genes are located near to a significantly associated variant, MAGMA assigns low p-values to all of them.
The Rare variant tab displays gene-level associations calculated from rare variants. The underlying data come from the following datasets:
- The AMP T2D-GENES T2D exome sequence analysis (T2D associations), published in Flannick et al. 2019. These associations were calculated for the gene overall (the canonical transcript) and for each individual transcript.
- The AMP T2D-GENES quantitative trait exome sequence analysis (23 cardiometabolic traits), published in Dornbos et al. 2022. These associations were calculated for the gene overall (the canonical transcript) and for each individual transcript.
- A pre-publication study (Jurgens SJ, Wang X, et al., Submitted, 2023) of rare-variant gene-level associations for 601 diseases across more than 600,000 individuals, based on exome and whole-genome sequence data. On the Gene page, associations from this study are displayed only for phenotypes that map exactly to Knowledge Portal phenotypes. These associations were calculated for only the gene overall (the canonical transcript).
- Gene-level association scores derived from the Genebass resource and based on exome sequencing data from the UK Biobank. Associations from this resource are displayed for 189 phenotypes that map exactly to Knowledge Portal phenotypes. These associations were calculated for only the gene overall (the canonical transcript).
For each gene, a burden test was performed to calculate a gene-level association score for each phenotype based on the associations of variants within the coding sequence. Tests were performed using different "masks" (criteria for grouping variants into sets by their predicted impact); the plot and table show the lowest p-value calculated across all the masks. See this page for more details about masks.
Using the menus above the plot, you can filter both the plot and table by entering a gene name or (for the common and rare variant associations tabs) by setting a p-value threshold for the associations displayed.
Top pathways
This table lists the pathways that are enriched for genes with the top gene-level association scores for this phenotype:
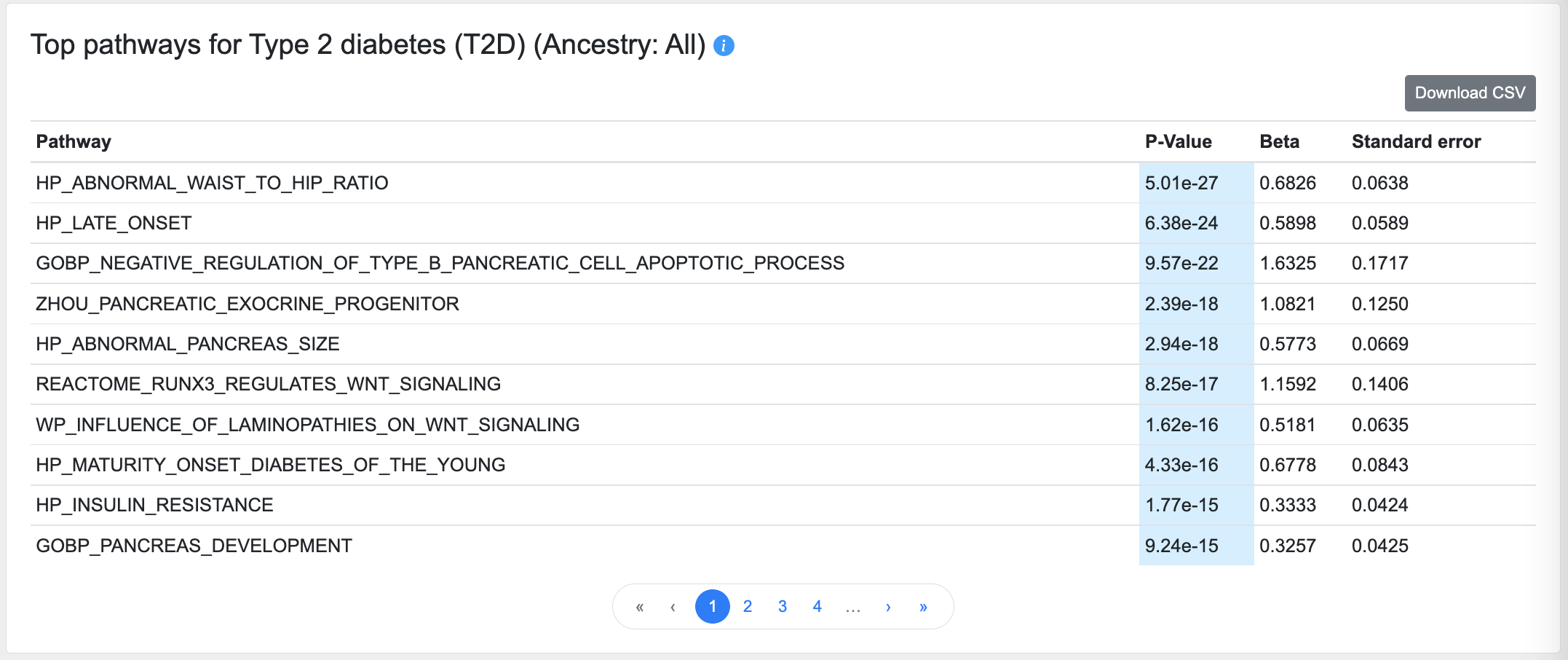
This calculation is made using MAGMA pathway analysis, using default parameters with the C2 (curated gene sets) and C5 (ontology gene sets) collections from the Molecular Signatures Database (MSigDB).
Genetic correlations
This table lists the phenotypes most closely correlated with the phenotype of the page, as calculated using cross-trait LD score regression:
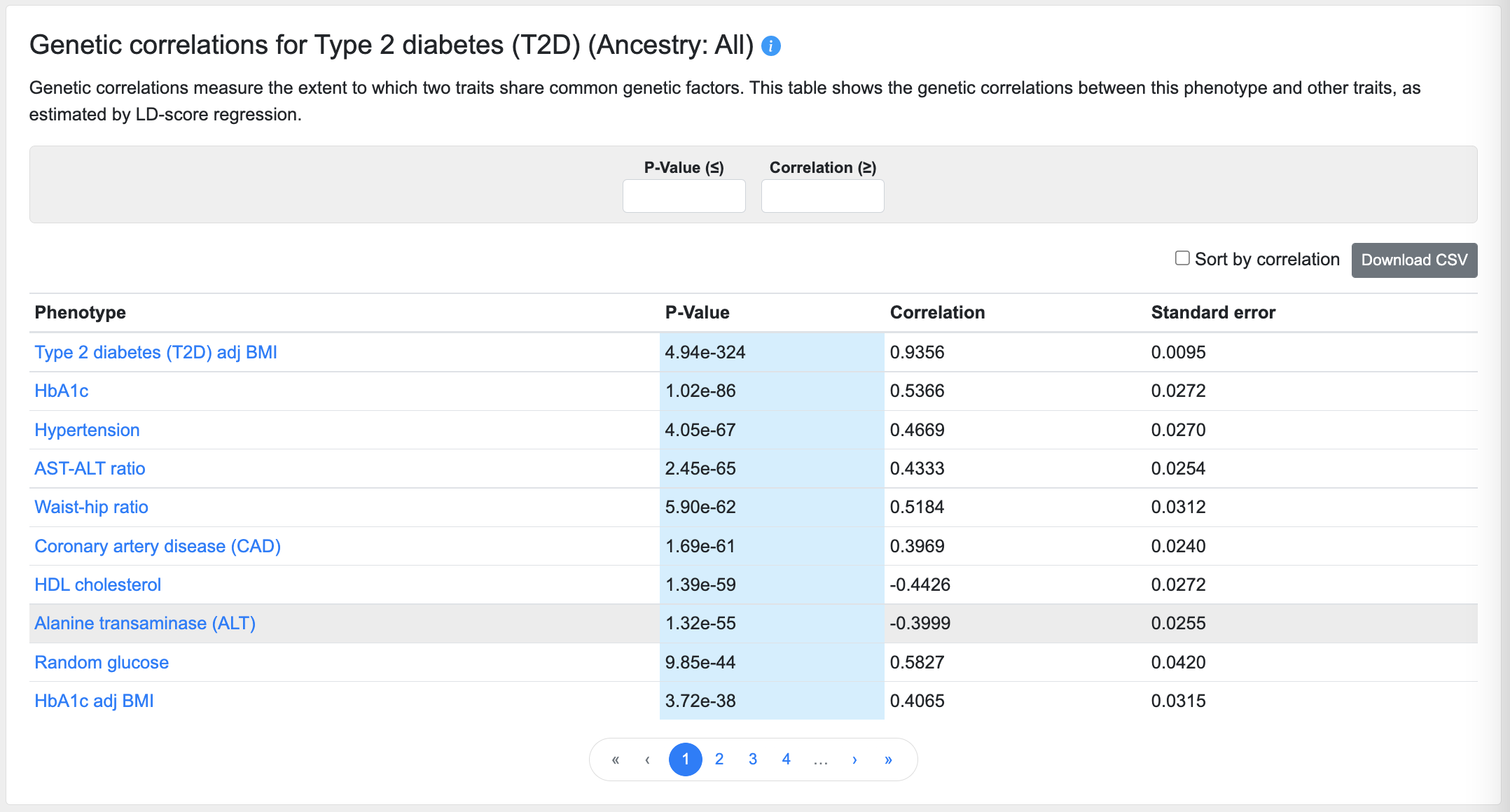
Using the filters above the table, you can enter a p-value (in the format "5e-8") to set an upper threshold for the correlations displayed, or enter a minimum correlation value threshold.
Globally enriched annotations
This table lists the annotation types and tissue groups that are enriched for genetic associations with the phenotype of the page, as determined using stratified LD score regression:
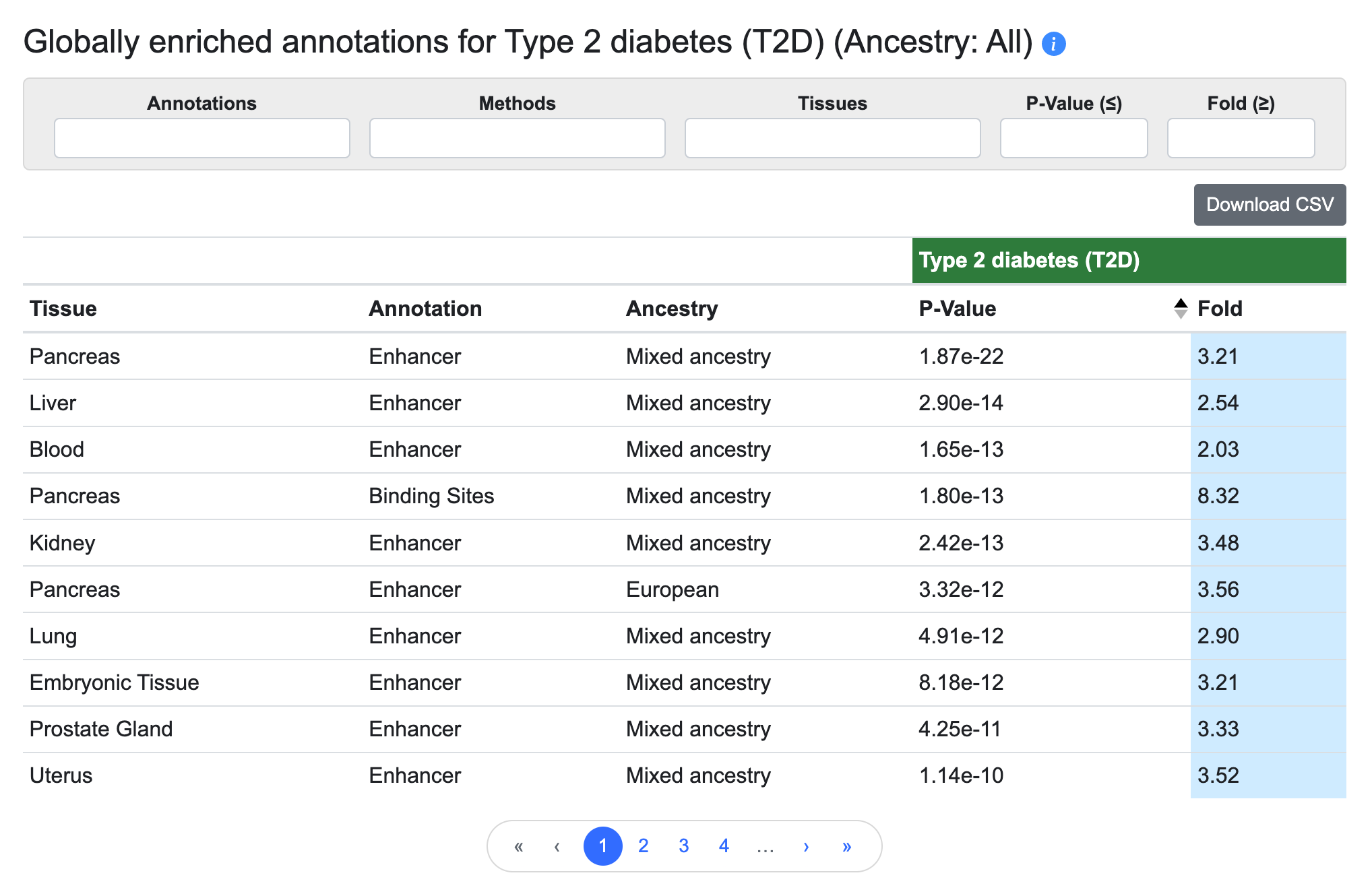
The annotations are derived from the Common Metabolic Diseases Genome Atlas (CMDGA). These enrichments can suggest which tissues are most relevant for a disease or trait.