The bNMF soft clustering approach categorizes genetic loci into groups representing likely disease mechanistic pathways.
Full documentation is available in:
Udler MS, Kim J, von Grotthuss M, Bonàs-Guarch S, Cole JB, et al. (2018) Type 2 diabetes genetic loci informed by multi-trait associations point to disease mechanisms and subtypes: A soft clustering analysis. PLOS Medicine 15(9): e1002654. https://doi.org/10.1371/journal.pmed.1002654. PMID: 30240442.
View a video from Hyungkyung Kim explaining how to navigate the Genetic Loci Clustering interfaces:
T2D Genetic Clustering
- Variant and trait selection
To obtain a comprehensive set of genetic variants associated with T2D, we started with the set of 88 variants reaching genome-wide significance aggregated by Mohlke and Boehnke 2015, and then added 37 additional loci that were reported in subsequent T2D large-scale genetic studies. We included 9 additional variants representing distinct signals at 6 loci (ANKRD55,DGKB, CDKN2A, KCNQ1, CCND2, and HNF4A). Of the 125 T2D variants considered, we selected a subset of 94 representative variants based on the condition that either the variant or a proxy of the variant had an association with T2D in the DIAGRAM version 3 (DIAGRAMv3) Stage 1 meta-analysis with P < 0.05. Proxies of variants were required to either be in linkage disequilibrium (r2 > 0.6) with an original T2D variant published at genome-wide significance, be the lead SNP at the T2D locus in DIAGRAMv3, or reach genome-wide significance in DIAGRAMv3. Additionally, variants (other than those representing distinct signals at a locus) were conservatively excluded if they fell within 500 kb of another variant on the list. Given that C/G and A/T alleles are ambiguous and can lead to errors in aligning alleles across GWAS, we avoided inclusion of ambiguous alleles, choosing proxies instead.
For the 94 T2D-associated variants, the T2D risk-increasing alleles were identified and clustering analysis was performed using the aligned T2D risk-increasing alleles. Summary association statistics for additional traits whose GWAS meta-analyses were publicly available were aggregated for each variant. These traits included glycemic traits from Meta-analyses of Glucose and Insulin-related traits Consortium (MAGIC), anthropometric traits from ANthropometric Traits (GIANT) consortium, visceral and subcutaneous adipose tissue, percent body fat and heart rate from VATGen consortium, additional birth weight and length from Genetic Investigation of Early Growth Genetics (ECG) consortium and serum laboratory values. To reduce noise, traits were only included if at least one variant was associated with the trait at a Bonferroni-corrected threshold of significance P < 5 x 10-4 (0.05/94).
- Bayesian non-negative matrix factorization (bNMF) clustering
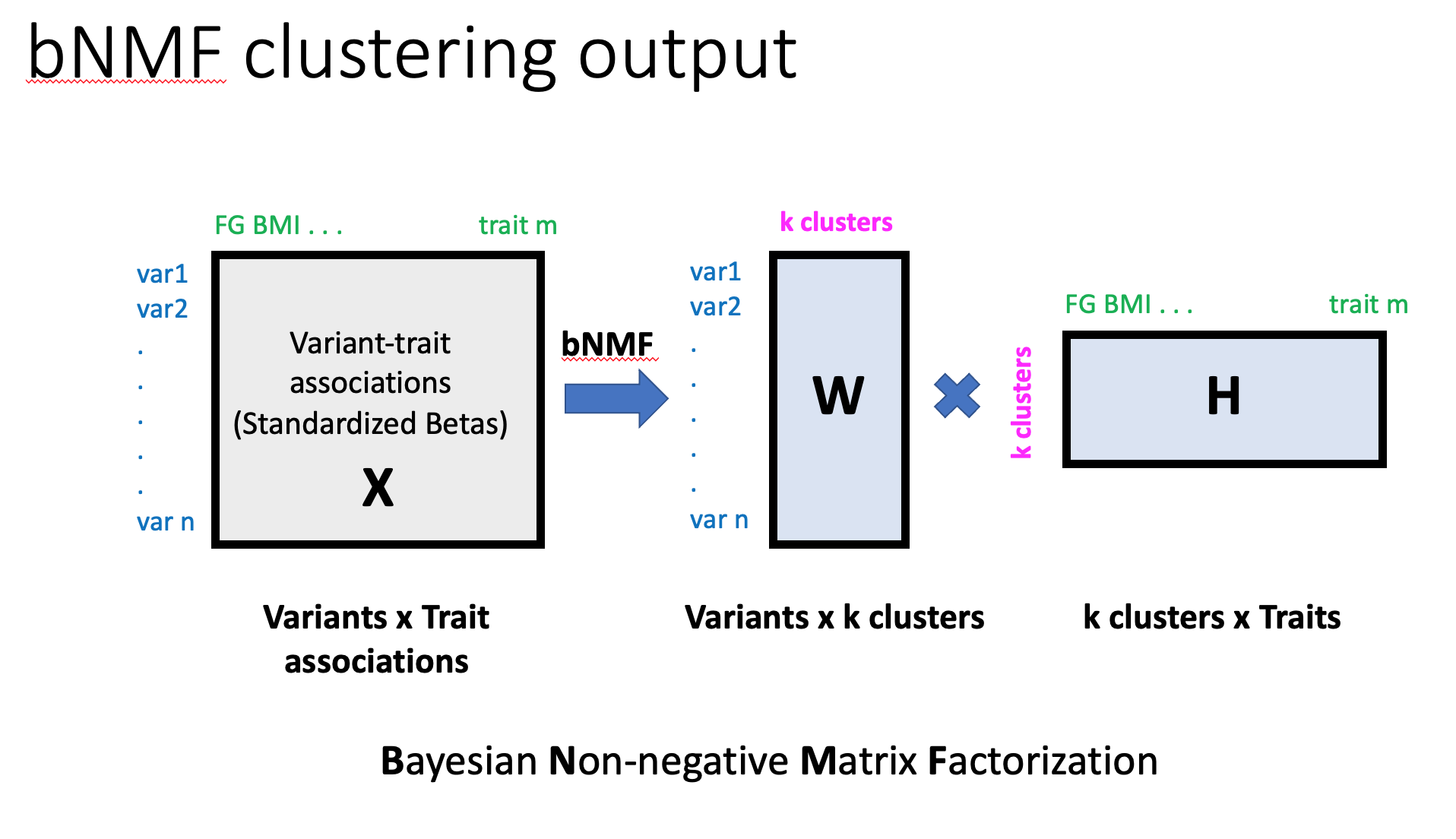
We generated standardized effect sizes for variant trait associations from GWAS by dividing the estimated regression coefficient beta by the standard error, using the GWAS summary statistic results. To address the marked differences in sample size across studies and allow for a more uniform comparison of phenotypes across studies, we scaled the standardized effect sizes by the square root of the study size, as estimated by mean sample size across all SNPs, forming the variant-trait association matrix Z.
To enable an inference for latent overlapping modules or clusters embedded in variant-trait associations, we modified the existing bNMF algorithm to explicitly account for both positive and negative associations. Each column of Z split into two separate entities: one contained all positive z-scores, the other all negative z-scores multiplied by −1, and setting zero otherwise, leading to the association matrix X comprised of doubled traits with positive and negative associations to variants.
Then, bNMF factorizes X into two matrices, W and HT, with an optimal rank K, as X ~ WH, corresponding to the association matrix of variants and traits to the determined clusters, respectively (see figure below). Determining the proper model order K is a key aspect in balancing data fidelity and complexity. bNMF is designed to suggest an optimal K best explaining X at the balance between an error measure, ||X−WH||2, and a penalty for model complexity derived from a nonnegative half-normal prior for W and H. The defining features of each cluster are determined by the most highly associated traits, which is a natural output of the bNMF approach. The results of clustering provide cluster-specific weights for each variant (W) and trait (H).