|
Inherited genetic variants linked to the presence or absence of a disease can reveal genes and pathways involved in the biological mechanisms underlying the disease. This understanding could in turn lead to more accurate prediction of risk and to better strategies for disease prevention and treatment. A forward genetics strategy for investigating a disease is to examine the genomes of people with or without the disease, looking for statistically significant genetic differences between them. These differences are referred to as variants or single-nucleotide polymorphisms (SNPs). This approach identifies disease-associated SNPs in an unbiased manner, with the goal of linking them to genes or regulatory pathways that affect the disease. A reverse genetics approach starts with a gene or genes hypothesized to be involved in the disease, based on previous evidence. For example, knockout of the gene might have led to a disease-relevant mouse phenotype. The researcher then investigates the phenotypic associations with variants affecting that gene, such as a variant predicted to introduce a premature stop codon. The goal of this approach is to investigate the function and physiological role of the gene product. |
|
In a forward genetics approach, the genomes of matched sets of individuals with a disease (cases) or without it (controls) are surveyed for SNPs with alleles that differ in frequency between the two groups. The frequency of each allele in people with disease is compared to its frequency in a comparable set of people without disease.
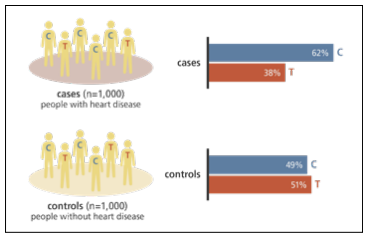
If there is a statistically significant difference (as shown by a p-value below a given threshold; see below) between the frequency of an allele in cases as compared to controls, the variant is said to be genetically associated with the disease. In the example shown, the C allele is found much more often in disease cases than in controls. The effect size or odds ratio for a variant (see below) measures the relative difference in frequencies; usually, for a fixed variant frequency, larger effect sizes lead to greater statistical association.
This association could mean that the C allele affects the function or regulation of a gene involved in the disease. Alternatively, it could merely be in linkage disequlibrium (see below) with a nearby variant that is in fact the causal variant. One limitation in forward genetics approaches is that correlations among variants mean that associations implicate the associated genomic regions, not individual variants.
Terminology used to describe variants
At any given nucleotide position in the human genome,
- The reference allele is the nucleotide that occurs at that position in the reference genome sequence (human genome build hg19).
- The alternate allele is a different nucleotide that occurs at that position in the genome of an experimental subject. Usually only one allele differing from the reference sequence is observed, but at some positions multiple differences are seen.
- The effect allele is the allele with which the size of the observed phenotypic effect is measured.
- The minor allele is the nucleotide that occurs least frequently at that position in the individuals who have been studied. Usually, but not always, the minor allele is the same as the effect allele. At some positions the reference allele is the minor allele, and populations of different ancestries may have different minor alleles.
Properties of variant alleles
- Each allele occurs in a population with a given frequency. The allele frequency is expressed as a fraction or percentage of chromosomes that carry the allele in the sampled population. For example, an allele with a frequency of 0.2 is present on 20% of the chromosomes in the population. Since individuals may be either homozygous for the allele, carrying two copies, or heterozygous, carrying one copy, the proportion of people carrying an allele differs from its allele frequency.
- Minor allele frequency (MAF) denotes the frequency of the minor allele in a given population. MAF is equivalent to the effect allele frequency when the minor and effect alleles are the same. Variants with MAF > 5% are considered to be common; variants with MAF between 0.05% and 5% are termed low-frequency; and variants with MAF < 0.05% are termed rare.
- Allele count refers to the number of chromosomes in the sampled population that carry an allele, and so reflects both allele frequency and sample size.
- Linkage disequilibrium (LD) is a measure of the extent of correlation between any two alleles. LD is usually expressed as r2, which is calculated using a formula that takes into account the frequency at which the alleles are found together on a single chromosome. An r2 value of 1 indicates that the alleles are completely correlated—that is, they are always inherited together. An r2 value of 0 indicates that the alleles are in linkage equilibrium—that is, they are inherited completely independently of each other.
- Variants with alleles that have strong phenotypic effects or that are predicted to cause severe molecular effects (for example, an allele that creates a translational stop codon within a gene, causing truncation of the encoded protein) are often referred to as “high-impact” variants.
Properties of genetic associations
- The p-value calculated for genetic associations represents the probability that the observed case/control frequency difference would occur by chance under the null hypothesis of no association between the SNP and disease: the lower the p-value, the greater the statistical significance of the association. A p-value of 5 x 10-8 or lower represents genome-wide significance; a p-value of 5 x 10-4 or lower represents locus-wide significance; and a p-value of 0.05 or lower represents nominal significance.
These significance cutoffs represent ways to account for the multiple testing in genome-wide studies. If a threshold of nominal significance (p = 0.05) is chosen for each test, then the threshold for N independent tests is 0.05/N. Since there are about 106 independent regions of the genome, a genome-wide association study (GWAS; see below) represents 106 independent tests, and genome-wide significance is 0.05/106 = 5 x 10-8.- Genome-wide significance is the “gold standard” for GWAS, because very few associations meeting this level of significance have been found to be false positives.
- Associations that do not meet the genome-wide significance threshold are not necessarily spurious. Obtaining additional data for replication may increase power, or integrating other types of data (for example, showing that a variant leads to loss of function of a gene product implicated in disease) could provide additional support for such associations.
- The genome-wide threshold of p < 5 x 10-8 that is appropriate for GWAS may not be stringent enough for sequence-based association studies: since many more variants are detected and tested, the number of independent tests is much greater.
- Once an association of genome-wide significance is observed between a variant and one trait, a p-value of 0.005 - 0.05 may be reasonable for associations of other variants in the region that are not in LD with the first variant, and thus could represent distinct causal variants.
- The odds ratio (OR) is used to represent the magnitude of the association between an allele and a binary, or dichotomous, disease or trait (e.g., T2D, ischemic stroke, chronic kidney disease), and it also represents the direction of its effect. An odds ratio near 1 means that the allele has little or no effect on disease; odds ratios greater than one mean that the effect allele is more likely to be carried by people with disease, and odds ratios less than one mean that the effect allele is more likely to be carried by people without disease, suggesting that it could be protective.
- Note that the numbers of cases and controls in a data set must be roughly equivalent in order to generate a meaningful OR.
- OR may not be accurate for variants with very low allele count in a particular data set.
- OR for the non-effect allele is equal to 1/OR of the effect allele.
- Effect size (beta) is analogous to the odds ratio but it can be applied to continuous (quantitative) traits such as BMI, fasting plasma glucose level, or cholesterol level. It represents the magnitude and direction of association between a variant and that trait. It is measured in units of population standard deviations; for example, a beta of 0.1 means that variant carriers have 0.1 standard deviation higher values than the mean. When a beta coefficient is given for a dichotomous trait, it represents the log(odds ratio).
- The confidence interval (CI) is a related to the certainty in the estimation of effect sizes that that accompanies odds ratios or effect sizes (beta). It represents an interval with a fixed probability of containing the true odds ratio or effect size. For example, 95% CI: (0.852 to 0.941) signifies that there is a 95% chance that the OR is between 0.852 and 0.941.
- Some studies generate a Z-score, which represents both the p-value and direction of effect of a variant-phenotype association.
- Some genetic associations are adjusted, meaning that a regression model is fit on one trait vs. another, the residuals are taken, and the association analysis is then conducted on the residuals. For example, to generate associations for HbA1c adjusted for BMI, each individual’s HbA1c levels are adjusted by BMI to generate the residuals—the level of HbA1c that is not explained by the regression on BMI—before the association analysis.
- The effective sample size of a study is defined as the number of samples in a balanced case:control design that would yield equivalent power as the original study. It is mathematically defined as 4 / (1 / n.case + 1 / n.ctrl).
Types of genetic association data
Knowledge Portals present results from multiple genetic association experiments that were performed using several different techniques. Each type of experiment reveals different aspects of disease genetics. They can be broadly divided into genotyping and sequencing studies.
Genotyping studies
Genome-wide association studies (GWAS) identify SNPs using microarrays that have been designed to detect hundreds of thousands of common genomic variants. Experimental subjects are classified by the presence of a disease and/or by various measurable traits such as fasting blood glucose, HDL cholesterol, or body mass index, and these measures are compared with the presence or absence of a variant allele, as described above.
Because large chunks of the genome tend to be inherited together and these haplotypes have been mapped, we can predict which nearby variants will accompany a common variant that is detected in a GWAS. This process of inferring association from linkage disequilibrium (LD; see above) is called imputation. Imputation can greatly increase the power and resolution of association studies.
An advantage of GWAS is that, as a well-established technique, experimental and statistical methods are standard and rigorous. And because GWAS look at common variants, association results usually apply across populations of different ancestry. Both of these factors contribute to good reproducibility of results between different studies.
Genetic associations detected in GWAS are expressed in terms of their p-value (the probability that the observed association would occur by chance) and either odds ratio (for binary traits) or effect size (for continuous traits). The significance of an association does not indicate the magnitude or direction of the effect of the variant on the trait: that is shown by the odds ratio or effect size.
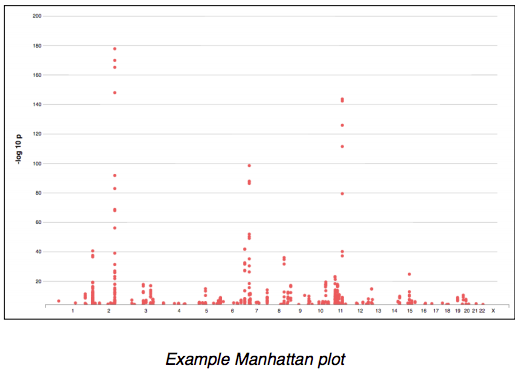
Overviews of GWAS data are displayed graphically in so-called Manhattan plots. The x-axis represents the genome as if all chromosomes were laid end-to-end, ordered by their chromosome number. Variants are plotted by the negative logarithm of the p-value for their associations with a specific trait (yaxis) vs. their chromosomal location (x-axis).
Using the Knowledge Portal Variant Finder tool, you can build a query for variants that are associated with multiple traits.
If apparently significant associations between SNPs and a phenotype are detected in an initial GWAS, those associations are often tested by replicating the analysis in an independent sample set. Another common way to pursue and confirm Example Manhattan plot results is to perform a meta-analysis that combines summary statistics from multiple studies, allowing associations to be tested across huge sample sizes.
GWAS data are the best source for comprehensive discovery of loci associated with diseases or traits. However, it is important to remember that a significant association between a common variant and a disease does not prove that the associated variant has any impact it. Rather, it indicates that the region of the genome around the common variant may contain a causal variant.
Other genotyping studies use the same technology as GWAS but survey selected subsets of the genome rather than the entire genome. Exome chip analysis uses an array that detects common variants found in the exome—that is, the part of the genome that encodes proteins. Other arrays have been designed to include sets of variants associated with specific processes of interest. For example, the Metabochip assays SNPs implicated in metabolic, cardiovascular, and anthropometric traits.
Fine mapping is an additional step performed to investigate in detail the region around a variant that is significantly associated with a phenotype. All of the genotyped and imputed variants near an association signal are tested for association, using high quality control and the largest possible sample sizes. The goal of fine mapping is to localize association signals to smaller and smaller sets of variants, so-called “credible sets,” to ultimately pinpoint the causal variant itself.
Sequencing studies
In contrast to genotyping studies, which assay the presence of a pre-determined set of variant alleles, sequencing studies determine the sequences of individual genomes. This means that they can detect extremely rare variants as well as common ones.
Whole-genome sequencing (WGS) is the most time-consuming and expensive method, but as its name suggests, it reveals every variant carried by each experimental subject. Whole-exome sequencing (WES), also referred to as exome sequence analysis, determines only the sequence of exons, the protein-coding regions of the genome. Since exons comprise only about 1.5% of the genome, exome sequence analysis is quicker and cheaper than whole-genome sequencing. However, it excludes from analysis any variants located in non-coding regions, such as those that affect transcriptional regulation.
Reverse genetics
A reverse genetics approach starts with a gene or genes suspected to have a role in a disease or trait, for reasons such as:
- A variant associated with the disease in GWAS lies near the gene.
- Variant alleles in a gene are associated with disease-related traits, such as levels of cholesterol or fasting glucose.
- A gene is hypothesized to be relevant to the physiology of the disease, from prior animal or cellular models.
Because there is a prior rationale for studying this gene, it may be justified to consider associations with less significant p-values than are considered in other approaches.
Best practices for using genetic association results in Knowledge Portals
- Use the Knowledge Portal as one of a suite of tools that can help generate or refine biological hypotheses. Results from analyses displayed on or conducted via the Knowledge Portal should not be taken as definitive on their own.
- See Dornbos et al. for a discussion of best practices while using the Knowledge Portals to evaluate genetic support for the role of a gene in a disease or trait.
- Be aware that the Knowledge Portal software is under active development and the data undergo periodic updates, so the results of any given analysis may change over time.
More information
Papers:
The Evolving Field of Genetic Epidemiology
Videos:
Molecular & Genetic Epidemiology
Epidemiological Studies - made easy!
Genetics Basics | Chromosomes, Genes, DNA
Genetics 101
P-values:
Khan Academy